中国抗癌协会
立即下载App《中国恶性肿瘤学科发展报告(2023)》——医学伦理学研究进展篇

1.概述
随着科技的飞速发展和医学领域的不断拓展,临床研究伦理审查作为保障研究参与者权益、确保科学研究合法合规进行的重要环节,越发凸显其在医学科学中的重要性。本报告将对我国临床研究伦理审查的现状、行业协会情况以及2023年度的进展进行全面分析和概述。
临床研究伦理审查的发展始于上世纪90年代,随着时间的推移,我国政府逐步完善了相关政策法规,确保医学研究符合伦理原则和法律法规。国际标准和指南,如世界医学大会赫尔辛基宣言,对临床研究伦理审查起到了重要推动作用,同时也促进了各国的监管法规和准则的制定,为临床研究的合法合规提供了坚实保障。
行业协会在临床研究伦理审查的学科发展中扮演着重要角色。中华医学会、中国抗癌协会等行业协会致力于提升行业标准与伦理素养,推动医学伦理的发展和实践。这些协会的不懈努力为医学研究提供了重要支持,促进了临床研究伦理审查的规范化和专业化水平的提升。
2023年度在临床研究伦理审查领域取得了新的进展。国家卫健委和国家药监局联合发布了新通知,进一步规范了临床研究者发起的伦理审查管理,为研究参与者的权益保护提供了更加明确的法律依据。同时,科技部等十部门也推出了《科技伦理审查办法(试行)》,提升了审查水平和效率,为临床研究的健康发展奠定了更加坚实的基础。
本报告将在接下来的章节中,对以上内容进行更详细的阐述和分析,以期为临床研究伦理审查的规范化和健康发展提供有益的参考和指导。

我国临床研究伦理审查的行业进展
我国临床研究伦理审查学科在国际标准指引下逐步完善,政府部门不断加强监管。行业协会包括中国抗癌协会等,致力于提升行业标准与伦理素养。2023年,国家卫健委和药监局联合发布新通知规范了临床研究者发起的伦理审查管理,科技部等十部门推出《科技伦理审查办法(试行)》,进一步提升了审查水平。01
临床研究的监管与政策环境新变化
以《世界医学大会赫尔辛基宣言》等为纲领的临床研究伦理审查国际标准和指南,在各国制定临床研究监管法规和准则,推动全球临床研究伦理审查的一致性以及在充分保护研究参与者的权益方面发挥着重要指导作用。
中国政府对临床研究伦理审查的政策法规监管最早可追溯到上世纪90年代,国家卫生部首次要求开展药物临床试验的临床药理基地必须成立伦理委员会。规定伦理委员会由5-7人组成,设置主任委员和副主任委员;申请注册临床试验的新药经卫生部批准下达临床试验任务给有关基地后,负责该项目的基地应与委托单位商定临床试验方案,交该基地医学伦理委员会审查,符合医学伦理要求并经医学伦理委员会主任委员签字后,方可组织参加试验的临床单位详细讨论方案,并按方案要求进行临床试验。1999年9月1日,由5位临床药理专家起草、国家药品监督管理局正式颁布的《药品临床试验管理规范》,开启了中国临床研究伦理审查政策法规监管的元年。规范特别强调所有以人为对象的研究必须符合《赫尔辛基宣言》和国际医学科学组织委员会颁布的《人体生物医学研究国际道德指南》的伦理原则,即公正、尊重人格、力求使研究参与者最大程度受益和尽可能避免伤害。其中“研究参与者的权益保障”专章对伦理审查委员会的人员组成、审查原则、审查流程、审查要点以及审查意见做出监管规定,并明确伦理委员会与知情同意书是保障研究参与者权益的主要措施。
进入本世纪,面对伦理学科及道德理念的不断演变和发展;临床研究前沿新技术和新疗法的持续涌现;信息化远程审查流程和电子伦理审查系统的逐渐应用;不同研究机构伦理审查结果互认机制的深入探索;全球性公共卫生事件对多中心临床研究伦理审查和研究参与者权益保护的冲击;临床研究伦理审查的政策法规监管必然要不断适应和解决由此产生的系列伦理难题。国家监管部门积极融入ICH全球监管体系,以修订《药品管理法》为引领,与时俱进地完善临床研究监管法规、指南和指导原则,其中涵盖的相关伦理审查章节和条款与国际准则全面接轨,对中国临床研究的质量和研究参与者权益保护起到了有效的监管作用,同时极大促进了中国临床研究行业的健康发展。
临床研究的范畴既包含了创新药物/器械的上市注册研究,也涵盖了研究者发起的优化患者诊疗新技术及新疗法的创新临床研究,如上市后药物/器械的临床评价和基于真实世界数据的前瞻性/回顾性临床研究。国家对前者的监管相对规范并逐渐形成体系,而对后者的监管尚处于起步阶段,在近十年先后出现贺建奎基因编辑违规、遗传资源泄露等典型事件,使涉及的伦理审查问题和隐患面临巨大挑战。国家卫健委和国家药品监督管理局采用联动监管机制,于近几年先后就研究者发起的临床研究、我国人类遗传资源管理等领域出台一系列新政,为医疗卫生机构开展研究者发起临床研究的伦理审查管理,以及站在国家战略安全高度规范管理研究参与者的遗传和隐私信息加强了监管机制。国家科技部等十部委也顺势而上,出台了科技伦理审查办法,为贯彻落实习近平总书记 “要前瞻研判科技发展带来的规则冲突、社会风险、伦理挑战,完善相关法律法规、伦理审查规则及监管框架。”的重要指示,创建我国促进科技创新、科技伦理与科技安全相统一,健全多方参与、协同共治的科技伦理治理体制的健康生态环境打下了坚实的基础。
经过三十多年的不懈努力,中国临床研究的监管与政策环境发生了可喜的变化,伦理审查理念和运行机制也拓展出崭新的局面。抗肿瘤药物/器械的创新临床研究以其前沿技术的创新应用,人文伦理的复杂多样性,不可置疑的引领和实践着临床研究伦理审查的挑战命题,必将通过研究从业者和研究参与者的共同努力,成长为模范遵循中国临床研究的伦理审查政策规范,以及最大限度保护研究参与者权益的行业典范。
02
临床研究伦理审查的相关法律法规
临床研究伦理审查必须遵守我国相关的法律、行政法规、部门规章等,遵守法律是临床研究伦理审查的底线和基础。
法律层面涉及《民法典》、《刑法》、《基本医疗卫生与健康促进法》、《科学技术进步法》、《生物安全法》、《个人信息保护法》等,2023年没有临床研究伦理审查相关法律颁布或修订。
行政法规层面:2023年12月颁布修订的《人体器官捐献和移植条例》,将《人体器官移植条例》名称改为《人体器官捐献和移植条例》,着重完善器官获取和分配制度,加强器官移植技术应用管理等。
部门规章层面:2023年2月教育部、国家中医药管理局、国家卫生健康委员会、科学技术部联合颁布《涉及人的生命科学和医学研究伦理审查办法》,《涉及人的生命科学和医学研究伦理审查办法》和2016年发布的《涉及人的生物医学研究伦理审查办法》(原卫生计生委令11号)主要制度框架、伦理审查方式、知情同意等总体上是一致的,在一定期限内,机构的具体伦理审查实践,可以以《涉及人的生命科学和医学研究伦理审查办法》作为指导;对医疗卫生机构伦理审查的违规行为,各级卫生行政部门可以11号令为依据进行处理。
2023年9月科技部、教育部、工业和信息化部、农业农村部、国家卫生健康委、中国科学院、中国社科院、中国工程院、中国科协、中央军委科技委发布《科技伦理审查办法(试行)》,该办法第五十四条:相关行业主管部门对本领域科技伦理(审查)委员会设立或科技伦理审查有特殊规定且符合本办法精神的,从其规定。而《药物临床试验质量管理规范》、《医疗器械临床试验质量管理规范》、《涉及人的生命科学和医学研究伦理审查办法》都是临床研究伦理审查的特殊规定,遵守即可。
2023年11月国家药监局颁布《药物临床试验机构监督检查办法(试行)》为进一步加强对药物临床试验机构的管理,规范药物临床试验机构监督检查工作。
2023年12月国家卫生健康委、国家中医药局、国家疾控局联合制定了《医疗监督执法工作规范(试行)》第二十条医疗技术临床应用管理监督执法:(六)查看临床研究项目的伦理审查管理、获取知情同意、费用收取、规范开展等情况等。
03
医学伦理审查委员会的基本情况(2023)
为了解我国肿瘤专业临床试验机构医学伦理审查委员会的实际工作情况,中国抗癌协会医学伦理学专委会抽取了8家肿瘤专科三甲医院和2家综合性三甲医院的10个伦理委员会的数据,覆盖东北、华北、西北、华中、华东、华南和西南地区。(见表1)
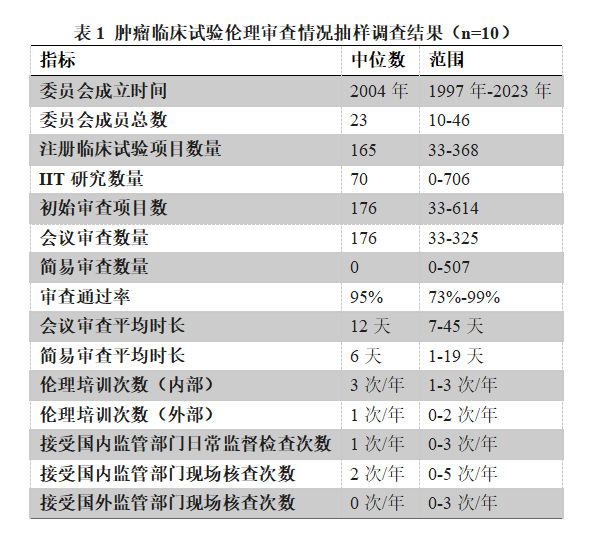
3.1 分析方法
对各委员会数据进行汇总、计算中位数并生成统计表
3.2 主要发现
(1) 委员会成立时间长短不一,成立最早的委员会于1997年成立,最晚的委员会于2023年成立。
(2) 委员会成员总数差异较大,范围为10-46人。
(3) 注册临床试验项目数量和IIT研究数量差异较大,分别为33-368项和0-706项。
(4) 初始审查项目数和会议审查数量差异较大,分别为33-614项和33-325项。
(5) 审查通过率总体较高,范围为73%-99%。
(6) 会议审查平均时长和简易审查平均时长差异较大,分别为7-45天和1-19天。
(7) 基本落实内部培训要求,但培训次数较少、培训内容实用性有待加强;大部分伦理委员会未组织外部培训,缺少与外有效交流不利于伦理审查理念与能力的同质化发展。
(8) 接受国内监管部门日常监督检查次数和接受国内监管部门现场核查次数差异较大,分别为0-3次/年和0-5次/年。
(9) 接受国外监管部门现场核查次数总体较少,范围为0-3次/年。
3.3 存在问题
(1) 部分委员会成员审查能力与水平不足,需加强法规和伦理培训。
(2) 部分委员会伦理审查流程不够完善,需进一步优化。
(3) 部分委员会伦理审查工作面临人员变动、预算不足等挑战。
3.4数据分析的局限性
本次分析仅基于10个委员会的数据,结果可能无法代表所有伦理审查委员会的情况。
04
国内临床研究伦理审查学术组织与社会团体发展动态
随着医疗技术的不断发展,临床研究伦理审查面临着日益复杂的伦理问题。政府及各界人士对如何更好地完成伦理审查以保护研究参与者权益十分关注。社会组织在这一进程中发挥着重要作用,通过整合资源,促进了临床研究伦理审查的发展。
4.1 已注册伦理相关社会组织
根据全国社会组织信用信息公示平台(https://xxgs.chinanpo.mca.gov.cn/gsxt/newList)的数据(截至2024年1月),已注册的伦理相关社会组织共有60家,其中正常运行的组织有49家,注销或撤销的有11家。这些组织的业务范围涵盖伦理道德类、伦理类、医学伦理类等,主要受到社会科学界联合会、科学技术协会、卫生健康委等部门的监管。
成立时间:关于这些组织成立时间统计显示,大多数伦理学会/协会成立时间较长,近年来成立的较少。其中,学术型组织居多,而针对医学伦理审查的社会组织较为稀少。(表2)
业务主管部门:不同社会组织的业务主管部门也呈现出一定的差异。以社会科学界联合会为主的社会组织最多,而医学伦理类的组织则以卫生健康委员会为主。这表明伦理学社团活动更多活跃于社会科学理论研究范畴,医学伦理领域学术组织数量较少,科技研发领域的伦理学研究明显缺乏,临床研究伦理审查领域缺乏学术交流平台。(表3)
区域性伦理学社团情况:伦理学会等社会组织,业务范围多以伦理理论为主,医学伦理审查社会组织仅分布于8个省级行政区,业务主管部门多以卫生健康委员会为主。(表4)
4.2 国家一级学/协会成立二级分支机构情况
除了注册的一级社会团体外,还有部分社会组织以二级分会或专业委员会的形式存在。在国内一些大型学会中,也成立了伦理专业分会或专业学组,如中华医学会医学伦理学分会、中国抗癌协会医学伦理学专业委员会、中国药学会药物临床试验伦理学研究专业委员会等。这些分会或委员会的成立有助于促进临床研究伦理审查工作的开展。(表5)
通过上表可以发现,医学类国家级社会组织,成立伦理的分会/专委会的相对较少,统计的16家社会组织中,仅5家有伦理方面的分会或专委会,占比为31.25%,其中3家是近5年成立的。
总的来说,临床研究伦理审查学术组织与团体在推动伦理发展、制定标准和规范审查方面发挥着重要作用。尽管目前医学伦理审查行业协会较少,但随着社会伦理意识的不断增强,我们有信心在未来进一步完善伦理审查的行业标准,发挥监督作用,促进伦理审查工作的持续发展。
国外临床研究伦理审查的学科进展
2023年,国际临床研究伦理审查方面取得进展。国际和地区伦理审查协会,如世界医学协会(WMA)、世界卫生组织卫生伦理及管理部门、国际医学科学组织理事会(CIOMS)等,持续推动伦理标准的制定和更新,确保医学研究符合伦理原则和法律法规。各协会加强认证体系建设,提升全球伦理审查专业化水平。同时,通过举办会议、开发数据库、出版期刊等形式,促进国际合作与交流,推动全球医学研究和临床试验的规范化和发展。这些努力有助于促进全球医学研究的伦理水平提升,保障研究参与者的权益和安全。
01
国际临床研究伦理审查指南和标准的最新进展
国际上,临床研究伦理审查的标准和指南主要由国际组织、各国政府以及专业机构共同制定和推动。随着全球化和国际合作的加强,国际临床研究日益增多,临床研究伦理审查法规和政策不断得以完善并随着医学和伦理学的发展而持续更新和修订。放眼全球,具体的临床研究伦理审查指南和标准可能因不同的国际组织、国家或地区而有所差异,但大多数都基于相似的伦理原则和核心要求,即保护研究参与者的权益和安全,促进科学研究的可信度和公众的信任。
近年来,多个国际组织和机构对临床研究伦理审查的指南和标准进行了更新和修订。这些更新旨在反映最新的伦理原则、实践经验和科学方法,以确保研究的伦理性和科学性。同时,随着生物技术、基因编辑、人工智能等新兴领域和技术的快速发展,这些新兴领域和技术带来了新的伦理问题,如数据隐私保护、算法偏见等。因此,国际组织和机构正在积极制定针对这些新兴领域和技术的伦理审查指南和标准,以确保研究的伦理性和可持续性、适应科技进步、促进国际合作与交流、提高伦理审查质量、应对法规变化以及提升公众信任,进而保证临床研究在全球范围内符合最高的伦理标准,同时也更加关注参与者的权益和保护。
概括来说,国际上临床研究伦理审查指南和标准的更新体现在多个方面,以下是一些关键领域的最新进展:
(1) 全球伦理审查标准的协调:随着国际合作的加强,各国和地区在伦理审查标准上的协调越来越重要。国际组织如世界卫生组织(WHO)和国际医学科学组织理事会(CIOMS)等正致力于推动全球伦理审查标准的统一和协调,以减少重复审查和跨国研究的障碍。
(2) 关注研究参与者的权益保护:伦理审查的核心是保护研究参与者的权益和安全。最新的伦理审查指南和标准更加强调对参与者的尊重、公正和透明对待。这包括确保参与者充分了解研究的目的、风险、受益等信息,并在自愿同意的基础上参与研究。
(3) 加强风险评估和伦理审查的透明度:随着对伦理审查要求的提高,风险评估和伦理审查的透明度成为重要关注点。最新的伦理审查指南和标准要求研究者和伦理委员会在审查过程中提供详细的风险评估报告,并公开审查过程和结果,以增加研究的可信度和公众的信任。
(4) 强化弱势群体的保护:弱势群体(如儿童、老年人、孕妇、精神疾病患者等)在临床研究中的保护一直是伦理审查的重点。最新的指南和标准强调对弱势群体的特殊关注,要求研究者和伦理委员会在审查过程中充分考虑他们的特殊需求和权益,并采取额外的保护措施。
(5) 利用现代技术提高伦理审查效率:随着科技的发展,现代技术在伦理审查中的应用越来越广泛。例如,电子化的伦理审查系统、人工智能等技术的应用可以提高审查的效率和准确性。最新的伦理审查指南和标准鼓励研究者和伦理委员会利用这些技术工具来改进审查流程和提高审查质量。
具体来说,以美国为例,美国在临床研究伦理审查方面一直走在国际前列,其指南和标准的进展体现了对伦理审查的深入理解和不断提高的要求,比如联邦法规第45章第46节(45 CFR 46)、第21章第56部分(21 CFR 56)、第21章第50部分(21 CFR 50)和国家卫生研究院(NIH)政策等。美国食品和药物管理局(FDA)在临床研究伦理审查方面一直在不断完善其指南和标准,以确保药物、生物制品和设备的安全性和有效性,同时保护参与者的权益。以下是FDA在临床研究伦理审查指南和标准的最新进展:
(1) 法规的更新与完善:随着医学科技的进步和社会的发展,美国在临床试验管理方面提出了更高的要求。例如FDA的生物研究监测项目(Bioresearch Monitoring Program,BIMO)旨在通过定期检查确保临床试验中研究参与者的权益得到保护,研究数据的真实性和可靠性。此外,FDA还公开了一系列的合规检查信息,包括483报告、警告信和被取消资格的临床研究者数据库等,这不仅提高了临床试验的透明度,还为行业内部提供了重要的合规指引。
(2) 强化伦理审查的透明度和一致性:FDA加强了伦理审查的透明度和一致性要求,确保研究者和伦理委员会遵循统一的伦理审查标准。这包括发布详细的指南和法规,解释伦理审查的流程和标准,以便各方能够明确理解和执行。
(3) 伦理审查范围的拓展:伦理审查的范围已经从传统的“在人体上”的研究扩展到“有关人的样本、医疗记录、行为等科研活动”。这意味着,即使研究活动不涉及直接在人体上进行,只要涉及人的生物样本或信息,都需进行伦理审查,显示了美国在伦理审查上的全面性和细致性。这种拓展确保了所有涉及人的研究活动都能得到适当的伦理审查,从而减少了潜在的风险和不当行为。
(4) 关注伦理审查的质量和效率:FDA致力于提高伦理审查的质量和效率,减少不必要的延误和重复工作。这包括推动伦理委员会之间的合作和信息共享,建立快速审查机制,以及优化伦理审查的流程和标准。
(5) 加强弱势群体的保护:FDA特别关注对弱势群体的保护,包括儿童、孕妇、老年人等。FDA发布了针对这些群体的特殊伦理审查指导原则,要求研究者和伦理委员会在审查过程中充分考虑这些群体的特殊需求和权益。
(6) 关注国际伦理审查的协调:随着国际多中心研究的增加,FDA加强了与国际合作伙伴的沟通和协调,推动全球伦理审查的一致性和互认。这包括参与国际伦理审查指南的制定和修订,以及与其他国家和地区的监管机构建立合作关系。
(7) 利益冲突管理:随着对利益冲突问题的日益关注,美国临床研究伦理审查指南和标准对利益冲突的管理要求也进行了更新。这包括要求研究者在研究申请中详细披露任何可能影响研究结果的利益冲突,并确保伦理审查委员会对利益冲突进行严格的评估和管理。这一措施有助于减少利益冲突对研究结果的影响,确保研究的公正性和可靠性。
欧盟在临床研究伦理审查方面的最新进展集中在数据保护、透明度、伦理审查、弱势群体保护以及可持续性和责任等方面,反映了欧盟对临床研究伦理问题的持续关注和对研究参与者权益保护的承诺。以下是欧洲药品管理局(EMA)在临床研究伦理审查方面的最新进展:
(1) 修订的伦理审查指南:2014年EMA人用药品临床试验法规Reg.(EU)No 536/2014 取代2001/20/EC指令,监管EMA临床试验法律。EMA定期更新其伦理审查指南,以反映国际最佳实践和伦理原则的最新发展。这些指南旨在帮助伦理委员会和研究者确保临床研究符合伦理要求,并保护参与者的权益和安全。
(2) 强化伦理委员会的角色:EMA强调伦理委员会在临床研究伦理审查中的重要性,并推动其角色和职责的明确和强化。这包括确保伦理委员会的独立性和公正性,以及提高其在审查过程中的透明度和效率。
(3) 关注全球伦理审查的协调:随着国际多中心研究的增加,EMA致力于促进全球伦理审查的协调和一致性。这包括推动采用共同的伦理审查标准和程序,以减少重复审查和提高审查效率。
(4) 加强对弱势群体的保护:EMA也特别关注对弱势群体(如儿童、孕妇、老年人等)的保护,强调在伦理审查中应充分考虑这些群体的特殊需求和权益。
(5) 数据保护和隐私:随着数字技术的进步,临床研究中产生的个人数据保护和隐私问题日益突出。为此,欧盟2018年5月25日实施了《通用数据保护条例》(GDPR),确立了严格的数据保护规则,要求研究者在处理个人数据时必须获得明确的同意,并确保数据的安全。此外,欧盟数据保护委员会2019年还发布了《关于临床试验条例与通用数据保护条例之间相互作用的问答》(EDPB 3/2019号意见),分析了临床试验中个人数据处理的合法性基础。
(6) 临床试验的透明度和可访问性:为了提高临床试验的透明度和公众对临床试验结果的访问,欧盟要求临床试验的结果必须在欧洲临床试验数据库(EudraCT)中注册和报告。此外,还推出了《欧盟临床试验指令》(EU Clinical Trials Regulation, CTR),该指令自2022年起生效,旨在通过一个集中的门户(EU Clinical Trials Information System, EUCTIS)提高临床试验的透明度。
(7) 持续的专业发展和培训:EMA重视伦理委员会成员和专业人员的专业发展和培训。通过提供培训课程和研讨会,EMA帮助伦理委员会成员提高伦理审查的能力和水平。
日本在临床研究伦理审查方面的最新进展体现在伦理审查范围的扩大、伦理审查指南的更新、伦理审查制度的完善,以及国际合作与共识的达成这几个方面,这些进展反映了日本对临床研究伦理问题的持续关注和对研究参与者权益保护的承诺。
(1) 伦理审查法规的更新:日本政府也在努力完善伦理审查制度,提高审查质量,并确保审查过程的独立性和公正性。例如,日本厚生劳动省就发布了一系列的指导方针,以帮助研究机构建立和维护有效的伦理审查系统,确保与国际最佳实践保持一致。
(2) 强化伦理委员会的作用:日本药品医疗器械局(PMDA)强调伦理委员会在临床研究伦理审查中的核心作用,推动其独立、公正、透明地运作。这可能包括改进伦理委员会的组成和运作机制,提高审查的专业性和效率。
(3) 关注弱势群体的保护:与FDA和EMA类似,PMDA也特别关注弱势群体的保护。它可能发布针对儿童、老年人、孕妇等特殊人群的伦理审查指导原则,以确保他们的权益得到充分保护。
(4) 促进国际协作与互认:在全球化背景下,跨国的临床研究越来越多,因此国际间的伦理审查合作与共识变得尤为重要。日本参与了多个国际性的伦理审查共识的制定,如国际医学科学组织理事会(CIOMS)的指南,以及其他国际性的伦理审查合作协议。
(5) 利用现代技术提高审查效率:PMDA也在不断探索利用现代技术提高伦理审查的效率和准确性。这可能包括采用电子化的伦理审查系统、利用大数据和人工智能等先进技术辅助审查等。
综上所述,随着科技的进步和全球化的发展,临床研究伦理审查指南和标准更新在近年来发展迅速。伴随着新兴领域和新技术如基因编辑、大数据、人工智能等的涌现,国际临床研究伦理审查将继续面临新的挑战和机遇。未来,我们可以期待更加完善、统一和高效的伦理审查指南和标准体系,以更好地保护研究参与者的权益并促进全球健康事业的发展。
02
临床研究伦理审查领域的国际学术会议和培训交流
2.1 世界医学会(WMA)《赫尔辛基宣言》修订会议
《赫尔辛基宣言(DoH)》是世界医学协会(WMA)颁布的一项重要政策声明,自1964年首次发布以来,经历了多次修订。在2022年4月的WMA理事会会议上,成立了一个专门的工作组,旨在对宣言进行最新的修订。这个工作组由美国医学会(AMA)领导,并获得了中华医学会的积极支持。修订工作的过程注重协作、透明和包容,旨在确保各方的意见得到充分考虑。修订工作组采取了为期两年的区域和专题会议的方式,邀请了当地和国际专家参与讨论,以确保新版宣言的全面性和适用性。这些会议包括了来自不同地区的专家,比如以色列、巴西、丹麦、日本、梵蒂冈、南非和德国。修订草案计划于2024年10月在芬兰赫尔辛基举行的WMA理事会和大会上进行审议和最终通过。更多信息请访问网站:https://www.wma.net。
2.2 医学科研公共责任组织(PRIM&R)年会
美国的医学科研公共责任组织(Public Responsibility In Medicine and Research,PRIM&R)成立于1974年,是一家国际知名的非营利组织。该组织致力于通过提供教育、会员资格和其他专业资源,确保医学研究符合最高的伦理标准。PRIM&R年度会议是国际医学科研伦理领域的重要学术交流平台。2023年的PRIM&R年会于12月3日至6日在美国华盛顿特区举行,主题为“新时代的新模式”。会议汇集了来自人类研究参与者保护、实验动物关怀领域的专业人士和机构领导,共同探讨了当前医学研究和监管中面临的复杂和挑战性问题。
年会围绕着多个主题展开讨论,包括政治与研究的冲突、国际实地研究的经验教训、研究高危行为人群时的伦理考量等。特别是针对动物护理、人类研究参与者保护等专业人士提供了独特的教育需求,并提供了一个跨学科交流的平台。
开幕式大会中,进行了一次特别专题研讨——是什么让你夜不能寐:研究的未来前沿。这是一个由临床试验和药物开发、人工智能、非人灵长类动物研究、科学劳动力发展等领域的国家专家组成的多元化小组就研究企业的发展轨迹以及伦理和监督界可能面临的挑战发表了他们的看法。此外,由发展生物心理学家 Allyson J. Bennett 博士主讲的主题演讲探讨了人类和其他动物之间的科学研究,以及它们在新知识产生和广泛受益方面的作用。
PRIM&R年会提供了丰富的学习和培训机会,涵盖了研究监管中的最佳实践和关键概念。与会者可以与专业社区建立联系,并与监管机构、行业专业人士和领域专家交流。明年的年会将在美国西雅图举行。更多信息请访问网站:https://primr.org/conferences。
2.3 美国生命伦理与人文协会(ASBH)年会
美国生命伦理与人文学会(ASBH)成立于1998年,是一个教育组织,旨在促进生命伦理学和人文学科领域的思想交流和合作。该协会的战略重点包括组织年会、提供伦理咨询资源、推动健康人文,以及倡导多元化、公平性和包容性。
第25届ASBH年会于2023年10月11日至14日在美国巴尔的摩举行,主题为“回顾过去,激发未来:为包容性公共话语创造空间”。年会试图重新思考生命伦理学和人文学科在社会中的角色,并探讨如何应对分裂政策以及如何与持不同观点的人进行诚实和公开的讨论。关键议题包括如何构建对话和辩论、征求主要利益相关者观点以及消除医疗保健服务中的偏见。
随着人工智能技术的迅速发展,我们面临着日益复杂的道德困境,需要重新思考个人和社会层面的人类意义。2024年第26届ASBH年会主题为“生而为人意味着什么?”将于9月在圣路易斯举行。该年会将深入探讨技术如何影响人类本性、如何承认医护人员和患者的人性,以及人类在更广泛环境中的角色。更多信息请访问网站:https://asbh.org。
2.4 其他重要会议
(1) 世界生命伦理学、医学伦理学与卫生法会议(World Conference on Bioethics, Medical Ethics and Health Law)
这是生命伦理领域的重要国际学术盛会,由世界医学会(WMA)核心伙伴组成的国际生命伦理主席团(ICB)主办。会议旨在提供一个国际性平台,让与会者就生命伦理学、医学伦理学和卫生法领域的60多个主题和分主题进行富有成果的科学讨论。来自五大洲的近300个成员组成的大学和医疗机构参与其中,中国成员占据了21个。
(2) 中美医师职业精神研讨会
该研讨会由北京大学医学部中美医师职业精神研究中心和北京大学医学人文学院主办。会议的焦点在于临床医生可能遇到的典型案例,包括生殖伦理、急诊伦理等,旨在探讨临床实践中的伦理难题。来自中外的学者、临床医生、伦理学专家、医院管理者等共同探讨医师道德对社会制度和治理的重要性。
(3) 遗传学和人类生殖新兴技术监管治理国际会议和数据正义及同一健康(One Health)国际会议
这次会议由香港大学医学伦理及人文学部(医学院)主办,旨在研究新兴医疗技术监管治理的性质和影响,重点关注生殖健康、基因组医学和抗菌素耐药性的最新发展。此外,还探讨了数字化趋势对同一健康倡议的影响,特别关注数字工具在解决抗菌药物耐药性问题中的应用和相关的技术和伦理障碍。
(4) 第17届世界生命伦理学大会(WCB)
这是由国际生命伦理学协会(IAB)主办的大型生命伦理学盛会,将于2024年6月在卡塔尔多哈举行。会议的主题是“宗教、文化和全球生命伦理学”,旨在探讨这些基本要素之间的错综复杂的关系。会议强调包容性,欢迎各种生命伦理学方法和其他主题的讨论,旨在促进全球生命伦理学学术领域的贡献和影响力的加强。
03
国际和地区性的伦理审查专业协会发展动态
医学科学技术必须符合人类根本利益,接受道德治理。20世纪40年代后,生命科学和生物医学技术发展引发广泛伦理讨论,促使公众意识提升,推动伦理委员会成立。伦理委员会依据伦理学原则,监督高新科技在医学中的应用,确保造福人类。随着科技进步和伦理意识提升,各国制定法规规范生物医学研究,推动医学伦理委员会发展。国际组织和地区协会成立,推动解决全球和地区卫生研究的伦理差异,促进医学伦理发展。
3.1 国际与地区伦理审查协会
(1) 世界医学协会 (WMA):WMA自1947年成立以来,致力于确保医生道德实践的高标准。核心文件包括《日内瓦宣言》《赫尔辛基宣言》和《国际医学伦理准则》,定期修订以保持相关性。WMA通过一系列宣言、决议和声明为全球医疗行业提供伦理和指导。
(2) 世界卫生组织卫生伦理及管理部门 (WHO-The Health Ethic and Governance Unit):WHO卫生伦理与管理部门是解决世界卫生组织活动引发的伦理问题的协调中心,涵盖资源分配、新技术、临床决策和公共卫生等领域的难题。该部门还支持成员国解决本国伦理问题,发布了一系列指导文件,包括《药物临床试验管理规范指南》和《评审生物医学研究的伦理委员会工作指南》。
(3) 国际医学科学组织理事会 (CIOMS):CIOMS是一个非政府、非营利性机构,旨在通过制定指南促进公共卫生的发展。其贡献在于发布了在各个生物医学领域应用伦理原则的国际准则,为世界各国提供了伦理咨询和指导。
(4) 国际人道主义和伦理联盟(IHEU):IHEU是人道主义者和伦理学家的国际学术团体,致力于推动人道主义网络的构建。通过影响国际政策,为世界提供人道主义视角。每4年举办一次联合大会,出版《国际人道主义者》和《人道主义书目》。
(5) 国际生物伦理学协会 (IAB):IAB促进了世界各地生物伦理学和相关领域的联系、网络和思想交流,鼓励生物伦理学研究、教学和实践。通过跨文化讨论和支持早期生物伦理学家,推动全球生物伦理学的发展。
(6) 亚洲生物伦理协会(Asian Bioethics Association):亚洲生物伦理协会是致力于处理生物伦理问题的综合性国际非营利组织,旨在促进国际伦理讨论。于1990年由Eubios伦理学研究所成立,分别位于新西兰和日本,之后在泰国曼谷设立第三个基地。协会通过与联合国、亚洲生物伦理协会等合作,培养自由思想者、激励青年领袖,并提供专业技能培训。
(7) 德国医学伦理委员会协会(Arbeitskreis Medizinischer Ethik-Kommissionen):1983年成立的德国医学伦理委员会协会是由52个公法设立的医学委员会联合组成,是一个正式认可的服务机构。协会致力于代表各伦理委员会的共同利益和立场,参与国内外政治辩论和政策制定。其13个工作小组关注法律变化,提供伦理指南和培训,并为跨部门利益相关者组织圆桌会议,高效解决问题。
(8) Ethox中心:是牛津大学的多学科生物伦理学研究中心,旨在提高医疗保健实践和医学研究的伦理标准。自1999年以来,它在英国国家医疗服务体系内积极发展,为卫生专业人员和研究人员提供伦理支持和建议,促进伦理标准的提高。通过举办培训、建立网络和提供资源等方式,为临床伦理委员会制定实用指南,推动伦理学在医学领域的应用。
(9) 肯尼迪伦理研究院(Kennedy Institute of Ethics,KIE):肯尼迪伦理研究院成立于1971年,是乔治城大学的实践伦理学中心,也是世界上最古老的学术伦理中心之一。通过创新教学和高影响力项目,肯尼迪研究院致力于回应当今紧迫的伦理问题。与佩莱格里诺临床生物伦理学中心、环境正义计划等合作,发展新的项目和活动,推动伦理学在各领域的发展。
3.2国际与地区伦理审查协会的作用
(1) 制定和更新国际伦理标准
国际与地区伦理审查协会致力于推动伦理审查标准化和规范化,确保医学研究符合伦理原则和法律法规。重要文件包括《日内瓦宣言:医生誓言》、《赫尔辛基宣言》和《国际医学伦理准则》。
• 《日内瓦宣言:医生誓言》概括了医生职业的基本伦理原则,定期修订以保持一致性。
• 《赫尔辛基宣言》针对医学研究中的伦理挑战,正在进行最新修订。
• 《国际医学伦理准则》概述了医疗行业的伦理道德原则和职责,已经进行了最近修订。
CIOMS制定了重要的国际伦理准则,如《研究机构良好治理实践国际指南》《患者参与药物的开发、监管和安全使用》和《涉及人的生物医学研究的国际伦理准则》。这些准则涵盖了科学研究、药物开发、人类研究参与等多个领域,为实践提供了指导。
(2) 建设伦理审查认证体系
常见的认证体系包括SIDCER和AAHRPP。SIDCER认证标准侧重于伦理委员会的审查能力,强调依法办事,按照标准操作程序进行审查。而AAHRPP认证则依据美国和国际伦理规则,考察更为全面细致的标准。
(3) 加强国际合作与交流
通过举办会议、开发数据库、出版期刊等形式,促进不同国家和地区的伦理审查实践经验的分享和交流。
• 世界卫生组织、Eubios伦理学研究所等组织举办各种国际会议和论坛,促进生物伦理领域的交流。
• WHO开发了多种数据库和网络,以支持全球伦理问题的处理和交流。
(4) 提升全球伦理专业化水平
伦理审查专业协会鼓励会员组织提升伦理审查的专业化水平,包括加强伦理委员会成员的培训和教育、制作培训手册、提供在线培训网络课程等方面。
• 各种机构和大学提供医学伦理课程和培训,如WMA的医学伦理课程和Ethox中心的教育课程。
• 乔治城大学肯尼迪伦理学院通过多种形式的课程、研讨会和在线资源,支持生物伦理学领域的学习和专业化。
总之,国际和地区伦理审查协会通过制定标准、建设认证体系、加强合作交流和提升专业水平,为全球医学研究和临床试验提供重要支持。
我国肿瘤临床研究参与者保护的挑战与对策
2023年,我国肿瘤临床研究参与者保护面临诸多挑战。特殊群体如儿童、孕妇、老年人等参与研究,其生理特点和心理状态复杂多变,需要针对性的保护和关怀。伦理审查应重点关注方案设计、知情同意、参与者选择和风险控制措施,确保其权益和安全。应建立健全的保险制度和赔偿责任,加强隐私保护,防止重复纳入和信息泄露。通过严格的伦理审查、完善的法律法规以及专业化的管理机制,促进肿瘤临床研究参与者权益得到充分保障,推动临床研究合规发展。
01
知情同意面临的挑战与对策
1.1 知情同意复杂性增加
(1) 研究内容的多元化:随着肿瘤临床研究的不断发展,研究内容也越来越复杂,涉及靶向/免疫药物联合多学科综合治疗、生物标志物外送中心实验室检测、影像学和病理学的中心评价、长时间的生存随访、疾病进展后继续治疗或交叉给药等等。这些复杂的研究内容给知情同意的解释和理解带来了新的挑战。
(2) 研究对象的特殊性:肿瘤患者群体存在多样性,包括晚期肿瘤患者、儿童肿瘤患者、精神障碍肿瘤患者、老年肿瘤患者、孕妇或哺乳期妇女肿瘤患者等等。这些特殊人群的知情同意能力可能存在差异,需要研究者采取更加谨慎的态度进行解释和沟通。
(3) 知情同意形式的多样化:除了传统的书面知情同意之外,电子知情同意、视频知情同意、动态知情同意等新的知情同意形式也逐渐应用于肿瘤临床研究中。这些新的知情同意形式需要研究者熟悉并掌握相关操作流程,确保知情同意过程的有效性。
1.2 知情同意过程的挑战
(1) 研究者解释不到位:部分研究者可能由于专业术语使用过多、语言晦涩难懂、解释内容不全面等原因,导致患者无法充分理解研究内容和风险。
(2) 患者理解能力有限:部分患者由于文化程度低、认知障碍、语言障碍等原因,可能无法完全理解知情同意书的内容。
(3) 患者自主权受到影响:部分患者可能由于经济困难、家庭压力、医患关系等原因,在参与研究时自主意愿受到影响,无法自主做出是否参与研究的决定。
1.3 知情同意伦理审查的挑战
(1) 审查内容复杂:肿瘤临床研究的知情同意伦理审查需要审查研究方案、知情同意书、伦理审查表等多个文件,内容复杂,需要伦理委员会成员具备专业知识和丰富的经验。
(2) 审查标准不统一:不同伦理委员会对知情同意的审查尺度和要求可能存在差异,导致审查结果不一致,影响研究的开展。
(3) 审查资源有限:伦理委员会成员数量不足、专业知识更新不及时等问题可能会影响审查效率和质量。
1.4 完善知情同意相关策略
(1) 优化知情同意书
• 语言通俗易懂:知情同意书应当使用通俗易懂的语言,避免使用专业术语,确保普通大众能够理解。
• 内容全面完整:知情同意书应当涵盖所有必要的信息,包括研究的目的、方法、风险、获益、替代方案、退出研究的权利等。
• 针对特殊人群提供个性化的知情同意书:对于晚期肿瘤患者、儿童肿瘤患者、精神障碍肿瘤患者等特殊人群,应当提供个性化的知情同意书,以便更好地满足他们的知情同意需求。
(2) 规范知情同意过程
• 研究者应当接受知情同意培训:研究者应当接受知情同意相关的培训,具备良好的沟通能力,确保能够有效地向患者解释研究内容和风险。
• 为患者提供充足的时间和机会阅读知情同意书:患者应当有充足的时间和机会阅读知情同意书,并与研究者进行充分的沟通,以确保他们能够理解研究内容和风险。
• 尊重患者的自主意愿:研究者应当尊重患者的自主意愿,不得强迫或欺骗患者参与研究。
(3) 加强知情同意伦理审查
• 制定统一的知情同意审查标准和操作规范:制定统一的知情同意审查标准和操作规范,确保审查的一致性和质量。
• 加强对伦理委员会成员的培训和管理:加强对伦理委员会成员的培训和管理,提高他们的专业知识和审查能力。
• 利用信息化技术,提高伦理审查效率:利用信息化技术,建立知情同意审查平台,提高。
02
高风险临床研究参与者的安全性风险评估
早期临床试验、细胞治疗、免疫疗法、基因治疗等肿瘤领域的高风险临床研究面临着复杂的挑战。针对这些挑战,伦理委员会需要加强审查工作,确保研究参与者的安全和权益得到充分保障。
2.1 早期临床试验
早期临床试验具有探索性目的,研究内容多变,风险较高且多元化。伦理委员会在审查时需要重点关注试验的科学价值、前期研究基础、研究设计、安全性监控等方面。试验方案应有完善的风险控制措施和数据监测计划,以确保研究参与者的安全。
2.2 肿瘤免疫治疗
免疫治疗的发展迅速,但也伴随着免疫相关不良事件(irAE)的风险。伦理委员会在审查时需关注免疫治疗产品可能引发的各种不良反应,并确保研究参与者充分了解潜在的风险。特别是对于存在潜在毒性风险的特殊人群,审查工作更应谨慎,确保其知情同意的有效性和安全性监测的实施。
2.3 免疫细胞治疗
免疫细胞治疗在临床试验中面临着挑战,包括细胞因子风暴、神经毒性等副作用。伦理委员会在审查时应重点关注研究方案的安全性评估和应对措施的有效性,确保研究参与者的安全。对于细胞治疗产品的存储、运输和使用等方面也需严格监管,以保证其疗效和安全性。
综上所述,伦理委员会在审查高风险临床研究时,需要高度重视研究参与者的安全和权益。审查过程中,应充分考虑研究的科学价值、风险评估、监测计划等方面,以确保研究的合理性和安全性。同时,持续监测临床试验的进行,及时应对可能出现的安全问题,保障研究参与者的利益。
03
关注特殊人群
2023年度在社会法领域出现了一系列重要变化,主要集中在特殊群体权益保障和临床研究伦理审查方面。社会法的第二部分主要涵盖了在劳动法和社会保障法之外的特殊群体权益保障法。这一部分的规范旨在体现社会法保护弱势群体的核心理念。通过特殊群体权益保护法律的制定,可以将社会的矫正思想置于自由主义平等思想之上,通过国家的干预和积极主动的作为,实现超越形式平等的更高层次的社会公平。特殊群体的权益保障法包括老年人权益保障法、未成年人保护法、妇女权益保障法等。这些特殊群体,如儿童、未成年人、老年人、妇女等,是社会法体系中最典型的特殊群体,将其纳入社会法体系与“社会法本质上就是一个弱势群体保护法”的核心理念高度契合。
《赫尔辛基宣言》中强调了纳入弱势群体作为研究参与者时必须满足的条件。这包括确保研究是在非弱势群体无法开展的情况下进行的,并且弱势群体参与研究应当能够切实获益。针对非完全民事行为能力人作为弱势群体的情况,应在试验方案中详细说明纳入他们的理由和采取的具体保护措施。在临床试验中,应当在科学的试验方案设计、完善的知情同意过程、有资质且经验丰富的研究者以及条件完备的医疗机构的基础上,才能有效保护这部分研究参与者的权益。
特别是在肿瘤研究中,涉及特殊人群的参与时,伦理审查尤为重要。儿童、孕妇和老年人由于身体状况和生理特点的不同,可能需要特别关注他们的权益和特殊伦理问题。伦理审查机构通常会对涉及特殊人群的研究进行严格的审查,以确保研究设计和实施符合伦理标准,保护参与者的权益和安全。在儿科临床试验中,伦理审查更是严格,需要建立与儿科临床试验相匹配的制度和流程,确保审查效率和研究参与者权益的保护。
儿童、孕妇、老年人等特殊群体参与临床研究,特别是肿瘤临床研究时,伦理委员会需要从以下几个方面进行重点审查:
方案设计: 伦理委员会应特别关注弱势群体的参与,并确保他们能够公平地参与临床研究。审查方案设计时,需要关注弱势群体的纳入和排除标准及理由,并要求研究者包括减轻不良事件的预案在内的完善方案。经费预算也应考虑到减轻风险措施所需的资源。
知情同意: 知情同意在临床试验中至关重要,研究参与者应亲自签署。伦理委员会要求研究者向参与者和家属解释临床试验的具体内容,消除恐慌心理,确保他们理解试验的意义和重要性,并自愿参加。
研究参与者选择: 伦理委员会要求研究者遵循公平选择原则,确保研究参与者能够自主选择是否参与临床试验。对于无法做出知情决定的弱势群体,如儿童、认知障碍者等,研究者需要特别关注其权益和保护。
风险控制和利益保障措施: 伦理委员会呼吁在风险较大的临床试验中建立强制保险制度,并确保研究者提供足够的赔偿和补偿责任。对于特殊群体的临床试验,应有更加细化和限定的保险和赔偿责任范围,以确保研究参与者的权益得到充分保障。
综上所述,伦理委员会在审查肿瘤临床研究时,需要全面评估干预措施的风险和潜在受益,并确保弱势群体的研究参与者能够在合理的风险受益比下参与研究。此外,还需要防止重复纳入研究参与者和隐私泄露,以确保其权益和安全。
04
生物样本管理
2023年,在生物样本管理领域,伦理审查体系进一步完善,促进了临床试验和医疗研究的规范开展。针对生物样本的取得和利用,更加严格的伦理原则和审查流程确保了样本提供者的知情权和隐私权得到有效保护。知情同意书的内容和签署过程得到进一步细化,保障了参与者的权益。同时,针对个体隐私和数据安全的关注,隐私保护措施得到加强,确保了生物样本和相关信息的安全使用。在利益共享方面,对知识产权的保护和利益分配原则的明确,促进了研究成果的公正分享和合理利用。2023年的医学伦理审查学科发展,为生物样本管理提供了更加健全的法律和伦理框架,推动了医学研究的持续发展。
4.1 伦理原则
生物样本管理应遵循伦理原则,其中包括尊重个体自主权、保护个人隐私、确保知情同意、最大限度地减少伦理风险等。生物样本的伦理审查应当遵守国家相关法律法规和条例,主要依据如下:
• 中华人民共和国生物安全法
• 中华人民共和国数据安全法
• 中华人民共和国个人信息保护法
• 中华人民共和国人类遗传资源管理条例
• 涉及人的生物医学研究伦理审查办法
• 涉及人的生命科学和医学研究伦理审查办法
• 药物临床试验质量管理规范
• 医疗器械临床试验质量管理规范
同时参考《赫尔辛基宣言》,遵循尊重样本提供者自愿、有益、不伤害和公正的原则。
4.2 生物样本的取得
生物样本的取得方式包括临床试验生物样本、诊疗剩余生物样本和生物样本库样本。
• 临床试验生物样本应获得知情同意,保护个人隐私,确保采集过程安全无损伤。研究人员应提供详细的信息,让参与者充分了解试验目的、风险和利益。
• 诊疗剩余生物样本也需要知情同意,并且样本的采集、储存和隐私保护应得到严格控制。研究者应告知患者样本使用的目的和可能的风险。
• 生物样本库样本需进行泛知情同意,并充分尊重提供者的意愿。研究人员应明确告知样本提供者样本的使用范围和可能的后果。
4.3 伦理审查
涉及生物样本的研究须在提交申请资料且经过伦理委员会审查同意后方能正式开展。审查要素包括样本采集依据、研究者资质、知情同意程序等内容。
4.4 知情同意
知情同意是保证研究参与者自愿参与临床试验的重要程序。在收集生物样本前,研究者应向提供者介绍研究相关情况,并确保其理解并签署知情同意书。对于无行为能力的样本提供者,应获取其法定监护人的知情同意。
4.5 隐私保护
保护样本提供者的隐私和个人信息安全是伦理审查的重要考量因素之一。研究人员应采取措施保护个体隐私,确保合法使用生物样本。此外,研究者应明确告知样本提供者相关身份鉴别记录的保密事宜,并保证在研究结果发布时样本提供者的身份信息仍然保密。
4.6 利益共享
利益共享应遵循公平、透明和互惠的原则,保障研究参与者、机构和研究人员的权益。对于由临床试验生物样本所产生的知识产权,应依法保护,并确保相关利益公正合理地分配。
生物样本管理的伦理审查是医学研究的重要环节,应遵循伦理原则,确保研究参与者的权利和隐私得到保护,推动医学研究的健康发展。
05
患者参与
2023年,医学伦理审查学科特别关注肿瘤药物临床试验中如何提升患者参与程度的几个关键问题。首先是试验中患者体验信息收集不足,导致数据质量下降。其次是部分项目在设计中未充分考虑研究参与者的负担,特别是对照组。另外,研究参与者及家属对信息反馈参与度不足,存在沟通障碍和教育培训不足的情况。最后,对创新技术相关的风险未充分告知,增加了患者负担。医学伦理审查学科将继续关注这些问题,以确保研究参与者的权益得到有效保护,并提供更准确和全面的数据支持。
5.1 研究参与者自报结果的收集和使用不足
2023年,在受试者参与方面,医学伦理审查学科的发展着重关注试验对研究参与者体验信息收集的不充分或关注度不够的问题。随着肿瘤患者生存时间延长,抗肿瘤药物长期应用的安全性变得更为重要。传统的疗效和治疗评价通常由外部第三方进行,但患者对于疾病的影响、治疗偏好、预期和需求,以及治疗带来的改善或负担有着最直接的感受。因此,患者报告结局(PRO)成为了重要的数据来源,包括自身健康状况、功能状态、生活质量以及对治疗的满意度等。PRO数据可以提供传统评价指标无法反映的患者治疗体验和临床获益信息。
然而,当前肿瘤药物的注册临床试验中,有些项目收集PRO数据存在一些问题。首先,有些项目未设计合适的问卷以收集体验信息,或者由于回答不及时、缺失等问题导致数据质量不佳。其次,部分项目问卷设计不够完善,使用多个量表时存在条目重叠问题,增加了患者的负担,降低了数据质量。此外,有些项目设计不够优化,问卷内容过多,导致答卷时间过长,可能影响患者的回答质量。还有一些项目未充分说明每次完成问卷所需时间及精力,在知情同意书中知情同意不充分,影响了研究参与者对面临负担的理解。
针对以上问题,2023年度医学伦理审查学科将继续关注并推动解决问卷设计不完善、数据缺失和收集不全等问题,提高研究参与者的参与体验,确保他们的权益得到有效保护,为肿瘤治疗的进一步研究提供更为准确和全面的数据支持。
5.2 部分试验设计对研究参与者负担缺乏考量
在药物临床试验的设计中,应该以患者为中心,充分考虑并收集他们对疾病和治疗的体验和需求。合理的研究设计应当是减轻患者负担的重要环节,同时也是保护患者权益的伦理要求。然而,在一些研究设计中存在着对研究参与者负担考量不足的问题。
举例来说,有些试验设计可能更专注于一组受试者,而忽略了对其他组别的关注。比如,一项新辅助/辅助治疗肿瘤的随机、双盲III期临床研究中,由于过度关注实验组的结局,而忽视了对照组的考量。结果导致对照组的受试者需要在相对较短的时间内接受过多的医学检查,增加了他们的负担和不适感。
另外,一些双盲设计的抗肿瘤药物临床试验在处理疾病进展时,未充分考虑到研究参与者的后续治疗需求。在这种情况下,研究者可能无法及时向患者提供关于他们曾接受过的治疗信息,从而延迟了他们接受新的治疗或参与其他临床试验的机会,增加了患者和家属的不安和精神负担。
近年来的研究表明,在中国进行的一些抗肿瘤药物临床试验中,存在着缺乏揭盲过程的现象。这意味着在试验进行过程中,疾病进展后的治疗情况可能无法及时被揭示,给研究参与者的后续治疗和关怀带来了不确定性,增加了他们的风险和负担。
综上所述,对于药物临床试验的设计,应当充分考虑研究参与者的负担,并建立合理的揭盲程序,以保护他们的权益和安全
5.3 缺乏研究参与者及家属参与的信息反馈
患者体验数据(Patient Experience Data, PED)是指患者、家属、监护者提供的关于疾病和治疗经验、观点和需求的信息。这些数据对于药物研发至关重要,但在一些情况下,研究者与患者及其家属之间存在沟通障碍,导致信息反馈的参与度不足。
首先,家属在患者治疗决策中发挥着重要作用,但他们与研究者之间存在着沟通不畅的问题。在中国文化中,患者通常与家人共同生活,而家属往往会在治疗决策中起到决定性作用。然而,目前的伦理规范并未明确要求在知情同意过程中让家属参与,这可能导致研究者无法获取到家属的宝贵意见和补充信息。
其次,对患者及其家属进行患者体验信息反馈的教育培训不足。一些患者由于对报告不良事件的认识不足,担心会被劝退出试验组,因此未能及时或者完整地报告不适症状。这表明研究者需要加强对患者及其家属的教育,让他们充分了解信息反馈的重要性和安全性。
另外,医患关系也会影响信息反馈的参与度。一些患者可能由于对医生的信任不足而不愿意分享治疗过程中的体验和观点。因此,建立良好的医患关系对于促进信息反馈的参与度至关重要。
5.4 IIT研究设计中增加患者负担的信息告知不充分
研究者发起的临床研究(Investigator-Initiated Trials, IIT)是指医疗卫生机构开展的,以个体或群体为研究对象的活动,不以产品注册为目的。然而,在一些IIT研究设计中,存在着未充分告知患者增加负担的问题。
首先,对于创新技术相关的IIT,研究设计的差异性较大,包括细胞治疗和联合治疗等方面的临床研究。在知情同意书中,除了共性条款外,大多数并未充分告知患者可能面临的增加负担,如细胞制备失败导致的重新制备风险、长期随访的需求等。这可能会给患者带来不必要的焦虑和不安。
其次,随着药物研发速度加快,越来越多的研究者在探索联合治疗方案。然而,这些联合治疗方案的安全性和有效性尚不确定,因此需要对患者进行充分的告知,并建立良好的监管机制以确保患者的安全和权益。
综上所述,研究者在进行临床研究设计时,应充分考虑患者和家属的意见和需求,同时对患者增加负担的风险进行充分告知,以保护患者的权益和安全。
5.5 针对当前存在的问题,医学伦理学专委会提出以下建议:
(1) 关注对研究参与者体验信息收集:
• 在审查方案时,重点关注问卷设计及收集患者体验的内容,评估其复杂性和完成时间,确保问卷设计优化,并在知情同意书中充分告知可能的负担。
• 对预取消生活质量问卷的项目进行审查,确保试验结果科学、伦理和可靠,包括考虑是否有替代方案评估患者生活质量。
(2) 针对部分项目方案对研究参与者负担考虑的不足:
• 重新评估研究设计,减少对患者的额外负担,例如延长评估间隔时间或使用非侵入性评估方法。
• 在知情同意书中明确说明评估频率和可能的负担,以增加透明度,便于患者做出治疗选择。
(3) 针对研究参与者家属的信息反馈参与度不足:
• 鼓励研究者教育患者及家属,强调其权益和被保护,鼓励他们积极报告不适,并通过多渠道与他们沟通,了解他们的症状与不适。
• 在临床试验结束后,与患者分享试验结果,并鼓励获得他们的反馈。
(4) 针对IIT研究设计中增加患者负担的问题:
• 仔细审查研究计划和设计,确保提供充分的理论和实证依据,以支持联合治疗方案的合理性和潜在益处。
• 在知情同意过程中,详细说明联合治疗研究的风险和潜在益处,确保患者充分知情并自主决定是否参与研究。
总之,伦理委员会应重视患者体验信息的收集,关注研究参与者的负担,促进家属的参与,以及保障患者在临床试验中的权益和安全。
06
研究参与者个人信息保护
2023年,随着《数据安全法》《个人信息保护法》等法规的实施,我国加强医疗数据安全和个人信息保护的步伐不断加快。现代信息技术的飞速发展使得医疗数据的收集、存储和共享变得更加便捷,但也带来了隐私保护的挑战。伦理审查在临床研究中的关键作用日益凸显,其着重关注个人信息保护的规范性和有效性,以确保在平衡各方利益的基础上最大限度地保护研究参与者的个人信息。伦理委员会在审查临床研究方案时应重点关注以下方面:
6.1 信息保护
在收集参与者信息时,特别是涉及个人身份和健康信息时,研究团队应遵循隐私保护的伦理原则。审查时,伦理委员会应确保知情同意过程充分告知参与者个人信息的收集、使用和保护措施,确保参与者理解并同意数据的使用目的和方式。此外,要求研究团队在数据处理和存储过程中采取适当的安全措施,如数据加密、匿名化等,以防止信息泄露。
6.2 数据安全
伦理委员会还应关注研究团队在数据安全管理方面的措施。这包括确保数据的采集、存储、传输和处理过程中符合相关法律法规和伦理要求。研究团队应制定数据管理计划,明确数据使用的权限和范围,并建立数据安全的监控和审查机制,及时发现和应对数据安全问题。
6.2 权益平衡
在保护个人信息的同时,伦理委员会还需考虑数据的合理使用和共享。在审查研究方案时,应评估数据共享对研究社会价值的贡献以及对参与者隐私的潜在风险,并确保研究团队在数据共享时采取适当的措施保护参与者隐私,如匿名化处理、访问控制等。
通过对个人信息保护的详细审查和监管,伦理委员会可以确保临床研究在遵循伦理原则和法律法规的基础上开展,保障研究参与者的权益和隐私安全。
【主编】
洪明晃 中山大学附属肿瘤医院
阎 昭 中国抗癌协会
【副主编】
鲍 军 江苏省卫生健康委员会
陈 震 复旦大学附属肿瘤医院
曹 烨 中山大学附属肿瘤医院
范 贞 北京市百瑞律师事务所
李 洁 北京大学肿瘤医院
李 宁 中国医学科学院肿瘤医院
梁茂植 四川大学华西医院
刘云鹏 中国医科大学附属第一医院
刘志敏 云南省肿瘤医院
罗素霞 河南省肿瘤医院
李坤艳 湖南省肿瘤医院
孙 健 中山大学附属肿瘤医院
唐 健 天津医科大学
王贵英 河北医科大学第三医院
王晓稼 浙江省肿瘤医院
周 宏 重庆大学附属肿瘤医院
朱 骥 浙江省肿瘤医院
【编委】(按姓氏拼音排序)
丁 晶 河南省肿瘤医院
韩 珂 中山大学附属肿瘤医院
黄 怡 北京诺诚健华医药科技有限公司
姜 琳 拜耳医药保健有限公司
廖红舞 北京大学肿瘤医院
刘小玲 浙江省肿瘤医院
曲 雁 河北医科大学第三医院
佟建华 中国医科大学附属第一医院
王雨萌 中国抗癌协会
吴大维 中国医学科学院肿瘤医院
徐伟珍 浙江省肿瘤医院
叶联华 云南省肿瘤医院
张玮静 复旦大学附属肿瘤医院
赵 青 江苏省肿瘤医院
参考文献(References)
[1]中华人民共和国卫生部, 《关于临床药理基地工作指导原则》, 1995.
[2]国家药品监督管理局, 《药品临床试验管理规范》, 1999.
[3]全国人大常委会, 《药品管理法》, 2019.
[4]中华人民共和国卫生部, 国家药品监督管理局, 《药物临床试验质量管理规范》, 2003.
[5]中华人民共和国卫生部, 国家药品监督管理局, 《药物临床试验质量管理规范》, 2020.
[6]中华人民共和国卫生部, 国家药品监督管理局, 《医疗器械临床试验质量管理规范》,2022.
[7]国家药品监督管理局, 《药物临床试验伦理审查工作指导原则》, 2010.
[8]国家卫生健康委员会, 《涉及人的生物医学研究伦理审查办法》, 2016.
[9]国家卫生健康委员会, 《涉及人的临床研究伦理审查委员会建设指南》, 2023.
[10]国家卫生健康委员会, 《医疗卫生机构开展临床研究项目管理办法》, 2014.
[11]国家卫生健康委员会, 《医疗卫生机构开展研究者发起的临床研究管理办法》, 2020.
[12]国家科技部等十部委, 《科技伦理审查办法(试行)》, 2023.
[13]刘艺迪;何辉;周刚;美国FDA药物临床试验研发主体合规检查信息公开情况的介绍和启示[J];中国新药杂志;2024年02期.
[14]李长青;张彦彦;姚佩颖;肖瑶;王雪云;高语晨;杨建红;关于国外伦理委员会管理制度研究[J];药学进展;2021年09期.
[15]NMPA. 国家药监局药审中心关于发布 《组织患者参与药物研发的一般考虑指导原则(试行)》的通告 (2022年第46号) [EB/OL]. (2022).
[16]FDA. Patient-Focused Drug Development Guidance: Methods to Identify What is Important to Patients and Select, Develop or Modify Fit-for-Purpose Clinical Outcome Assessments [EB/OL]. (2022).
[17]FDA. Patient-Focused Drug Development: Collecting Comprehensive and Representative Input [EB/OL]. (2020).
[18]FDA. Patient-Focused Drug Development: Selecting, Developing, or Modifying Fit-for-Purpose Clinical Outcome Assessments Guidance for Industry, Food and Drug Administration Staff, and Other Stakeholders [EB/OL]. (2022).
[19]ICH. ICH E8(R1): GENERAL CONSIDERATIONS FOR CLINICAL STUDIES [M].
[20]NMPA. 国家药监局药审中心关于发布《以临床价值为导向的抗肿瘤药物临床研发指导原则》的通告(2021年第46号) [EB/OL]. (2021).
[21]NMPA. 国家药监局药审中心关于发布 《以患者为中心的药物临床试验设计技术指导原则(试行)》《以患者为中心的药物临床试验实施技术指导原则(试行)》《以患者为中心的药物获益-风险评估技术指导原则(试行)》的通告(2023年第44号) [EB/OL]. (2023).
[22]Dawei W, Shuangman M, Huiyao H, et al. Unblinding at disease progression in double-blinded randomized controlled cancer drug clinical trials: A controversy requires more attention [J]. Frontiers in Medicine, 2022, 9.
[23]Hobbs G S, Landrum M B, Arora N K, et al. The role of families in decisions regarding cancer treatments [J]. Cancer, 2015, 121(7): 1079–1087.
[24]洪明晃, 阎昭. 伦理审查-中国肿瘤整合诊治技术指南(CACA)[M].
[25]Zhou H, Yao M, Gu X, et al. Application of Patient-Reported Outcome Measurements in Clinical Trials in China [J]. JAMA Network Open, 2022, 5(5): e2211644.
[26]FDA. Placebos and Blinding in Randomized Controlled Cancer Clinical Trials for Drug and Biological Products Guidance for Industry [EB/OL]. (2019).
[27]李宪辰,康玫,邱燕,等.研究者发起的临床研究中研究参与者个人隐私保护探讨[J].中国医学伦理学, 33(12):1459-1462.
[28]药物临床试验 研究参与者隐私保护广东共识(2020年版).
[29]黄樱硕,张子龙,吴小芳,等.药物临床试验研究参与者隐私保护的有关伦理问题及其研究进展[J].中国医学伦理学, 33(9):1046-1052.
[30]李雪迎,王熙诚,沙若琪,等.真实世界数据研究的信息安全挑战[J].中国食品药品监管, 10(225):46-53.
[31]刘丹,周吉银.临床科研项目研究参与者隐私保护的伦理审查[J].中国医学伦理学,34(10):1306-1310.
[32]科技教育司. 关于印发涉及人的生命科学和医学研究伦理审查办法的通知[EB/OL].(2023-02-27).
[33]中国医院协会. 涉及人的临床研究伦理审查委员会建设指南(2023版)[EB/OL].(2023-06-28).
[34]国家科技部. 科技伦理审查办法(试行)[EB/OL]. (2023-09-07).
[35]巩琦凡,郑晓飞,付汉江.CRISPR基因编辑技术的发展及应用[J].中国生物化学与分子生物学报,2023,39(03):332-340.
[36]孙海波.基因编辑的法哲学辨思[J].比较法研究,2019(06):105-120.
[37]王浩东.基因编辑技术伦理风险的法律治理[J].锦州医科大学学报(社会科学版),2021,19(05):10-14.
[38]中国科学院.人类基因编辑研究报告全球发布提出科学、伦理与监管基本原则[EB/OL].(2017-02-15).
[39]孙那,王雅洁.生殖系细胞基因编辑的法律规制进路[J].中国医学伦理学,2023,36(12):1342-1349.
[40]陶林.论医学人工智能的运用、伦理风险与规制[J].青岛科技大学学报(社会科学版),2022,38(02):63-69.
[41]叶卓俊, 沈艳丽, 江晓, 袁蕙芸. 医学人工智能领域伦理治理重点研究[J]. 中国医学伦理学, 2024, 37 (01): 39-44.
[42]国家药品监督管理局医疗器械技术审评中心. 国家药监局器审中心关于发布影像超声人工智能软件(流程优化类功能)技术审评要点等4个审评要点的通告. [EB/OL]. (2023-07-10).
[43]丁瑜. 案例分析法在医学伦理学课程中的运用[C]//榆林市医学会.第二届全国医药研究论坛论文集(一),2023:6.
[44]张新庆.临床伦理学:新时代呼唤新作为[J].协和医学杂志,2020,11(05):638-640.
[45]王晓敏,刘星,周岚等.新医科视域下医学伦理学教学改革思考[J].中国医学伦理学,2021,34(10):1371-1375.
[46]袁曦.恶性肿瘤治疗中的伦理问题与确证[J].肿瘤代谢与营养电子杂志,2014,1(02):11-14.
[47]Cheng, N, Cui, X, Chen, C, et al. Exploration of Lung Cancer-Related Genetic Factors via Mendelian Randomization Method Based on Genomic and Transcriptomic Summarized Data. Front Cell Dev Biol. 2021; 9 800756.
[48]何玲玉,王玥,李闪闪等.临床研究之伦理治理框架: Emanuel八个“伦理原则”的审辨[J].医学与哲学,2019,40(16):1-5.
[49]田野,刘霞.基因编辑的良法善治:在谦抑与开放之间[J].深圳大学学报(人文社会科学版),2018,35(04):106-115.
[50]国家卫生健康委,教育部,科技部,国家中医药局,《涉及人的生命科学和医学研究伦理审查办法》,2023.