中国抗癌协会
立即下载App中国急性早幼粒细胞白血病诊疗指南(2018年版)
急性早幼粒细胞白血病(APL)是一种特殊类型的急性髓系白血病(AML),绝大多数患者具有特异性染色体易位t(15;17)(q22;q12),形成PML-RARα融合基因,其蛋白产物导致细胞分化阻滞和凋亡不足,是APL发生的主要分子机制[1,2]。APL易见于中青年人,平均发病年龄为44岁,APL占同期AML的10%~15%,发病率约0.23/10万[1]。APL临床表现凶险,起病及诱导治疗过程中容易发生出血和栓塞而引起死亡。近三十年来,由于全反式维甲酸(ATRA)及砷剂的规范化临床应用,APL已成为基本不用进行造血干细胞移植即可治愈的白血病[3,4]。
1.病史和体检
2.血液检查:血常规、血型,外周血涂片,生化,DIC相关指标检查,输血前有关传染性病原学检查。
3.骨髓检查:
(1)细胞形态学和组织化学:
以异常的颗粒增多的早幼粒细胞增生为主,且细胞形态较一致,胞质中有大小不均的颗粒,常见呈柴梱状的Auer小体。FAB分型根据颗粒的大小将APL分为:①M3a(粗颗粒型);②M3b(细颗粒型);③M3c(微颗粒型):较少见,易与其他类型AML混淆。细胞化学:APL的典型特征表现为过氧化酶强阳性、非特异性酯酶强阳性且不被氟化钠抑制、碱性磷酸酶和糖原染色(PAS)呈阴性或弱阳性。
(2)免疫分型:
免疫分型在APL诊断中起到辅助作用。其典型表现:表达CD13、CD33、CD117和MPO,不表达或弱表达CD34、HLA-DR、CD11b、CD14、CD64、CD56。少数表达CD56患者提示预后较差。
(3)细胞遗传学:
典型APL表现为t(15;17)(q22;q12)。变异型APL占2%,如t(11;17)(11q23;q12)、t(5;17)(5q35;q12)、t(11;17)(q13;q21)、der(17)、t(17;17)(q24;q12)、t(4;17)(q12;q21)、t(X;17)(p11;q21)、t(2;17)(q32;q21)、t(3;17)(q26;q21)、t(7;17)(q11;q21)、t(1;17)(q42;q21)等。5%的APL患者核型正常。常规染色体检测有时还可发现除t(15;17)以外的附加染色体异常。
(4)分子生物学:
①PML-RARα融合基因:98%以上的APL患者存在PML-RARα融合基因,另有低于2%的APL患者为其他类型融合基因(见以下变异型APL诊断标准),检测PML-RARα融合基因是诊断APL的最特异、敏感的方法之一,也是APL治疗方案选择、疗效评价、预后分析和复发预测最可靠的指标。实时定量PCR(RQ-PCR)可在99%的典型APL患者中检出PML-RARα融合基因,但仍有1%的APL患者可出现假阴性。②基因突变:部分APL患者可伴有FLT3-ITD突变。
4.其他检查:心电图,超声心动图(必要时),胸片,腹部B超或CT(必要时)。
5.如外周血血小板计数及纤维蛋白原定量明显下降,存在严重出血倾向时,则不建议行PICC插管。
1.FAB分型为AML-M3。
2.WHO 2016年分型为伴重现性遗传学异常急性髓系白血病亚型下的APL伴PML-RARα阳性。
3.t(15;17)APL的诊断标准:PML-RARα融合基因阳性或染色体/FISH证实t(15;17)(q22;q12)时可确诊。
4.变异型APL的诊断标准:具有APL的临床特征、细胞形态学表现,细胞遗传学或分子生物学检测发现t(11;17)(11q23;q12)/PLZF-RARα、t(5;17)(5q35;q12)/NPM-RARα、t(11;17)(q13;q21)/NuMA-RARα、der(17)/STAT5b-RARα、t(17;17)(q24;q12)/PRKAR1A-RARα、t(4;17)(q12;q21)/FIP1L1-RARα、t(X;17)(p11;q21)/BCOR-RARα、t(2;17)(q32;q21)/OBFC2A-RARα、t(3;17)(q26;q21)/TBLR1-RARα、t(7;17)(q11;q21)/GTF2I-RARα、t(1;17)(q42;q21)/IRF2BP2-RARα、t(17;17)(17q21;q12)/STAT3-RARα[5,6,7]。
流程见图1。
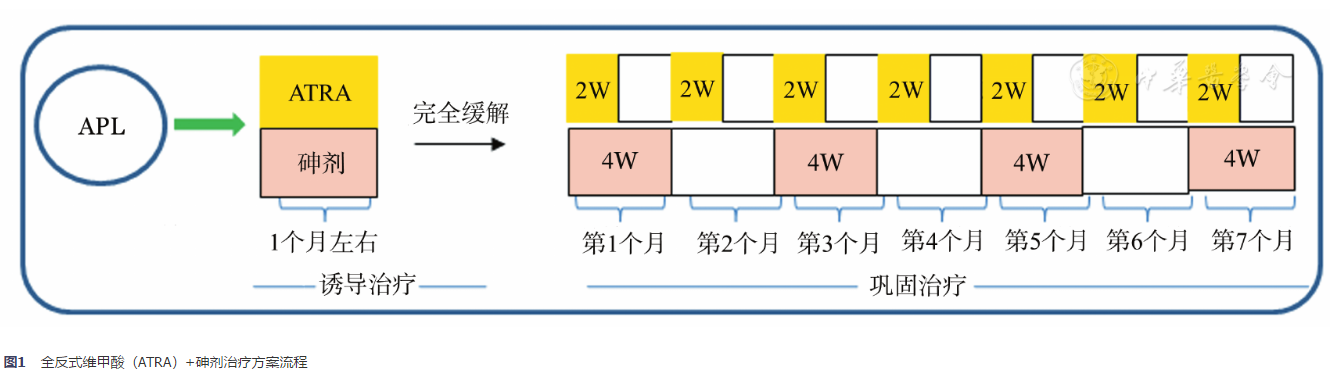
ATRA 25 mg·m-2·d-1同时联合三氧化二砷(简称亚砷酸)0.16 mg·kg-1·d-1或复方黄黛片60 mg·kg-1·d-1,直到完全缓解(CR),总计约1个月[治疗前WBC(4~10)×109/L,予以羟基脲1.0 g,每日3次,口服,应用天数按白细胞计数而定;治疗前WBC<4×109/L,待治疗中WBC>4×109/L时加羟基脲1.0 g,每日3次,口服,应用天数按白细胞计数而定;治疗中WBC>10×109/L时,酌情加用蒽环类药物或阿糖胞苷(Ara-C)]。
ATRA 25 mg·m-2·d-1×2周,间歇2周,为1个疗程,共7个疗程。亚砷酸0.16 mg·kg-1·d-1或者复方黄黛片60 mg·kg-1·d-1×4周,间歇4周,为1个疗程,共4个疗程。总计约7个月。
流程见图2。每3个月为1个周期。第1个月:ATRA 25 mg·m-2·d-1×2周,间歇2周;第2个月和第3个月亚砷酸0.16 mg·kg-1·d-1或复方黄黛片60 mg·kg-1·d-1×2周,间歇2周。完成3个周期,维持治疗期共计约9个月。
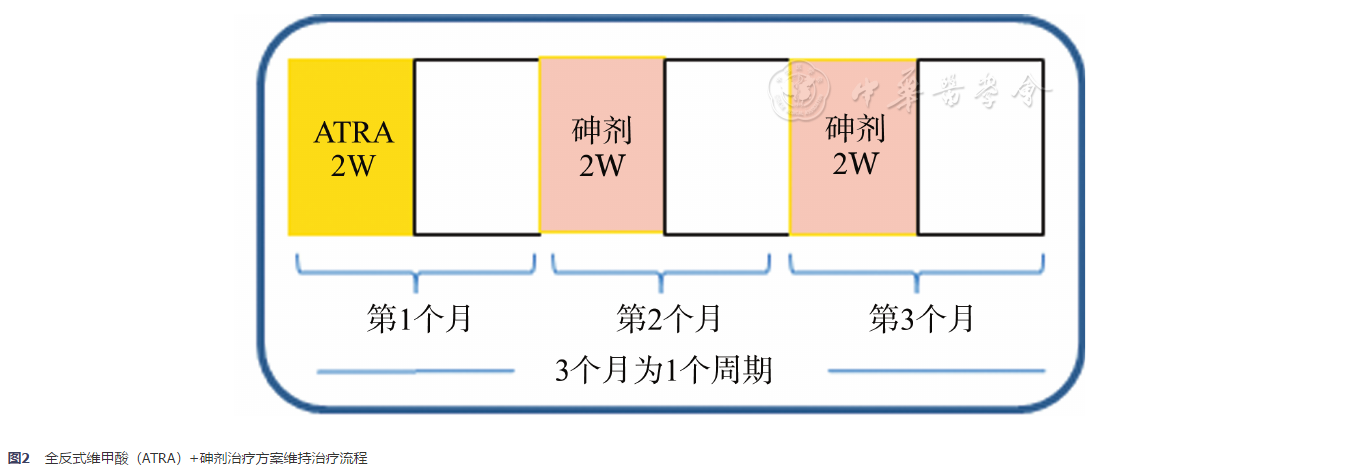
本文中亚砷酸均为静脉滴注。复方黄黛片(主要含四硫化四砷的复方制剂)及ATRA均为口服。

ATRA 25 mg·m-2·d-1联合亚砷酸0.16 mg·kg-1·d-1或复方黄黛片60 mg·kg-1·d-1,直到CR;蒽环类或者蒽醌类药物控制白细胞增高。
可选方案:
①HA方案:高三尖杉酯碱(HHT)2 mg·m-2·d-1,第1~7天;Ara-C 100 mg·m-2·d-1,第1~5天。
②MA方案:米托蒽醌(MIT)6~8 mg·m-2·d-1,第1~3天;Ara-C 100 mg·m-2·d-1,第1~5天。
③DA方案:柔红霉素(DNR)40 mg·m-2·d-1,第1~3天;Ara-C 100 mg·m-2·d-1,第1~5天。
④IA方案:去甲氧柔红霉素(IDA)8 mg·m-2·d-1,第1~3天;Ara-C 100 mg·m-2·d-1,第1~5天。
若第3次巩固化疗后未达到分子学转阴,可加用IDA(8 mg·m-2·d-1,第1~3天)和Ara-C (1.0 g/m2,每12 h 1次,第1~3天)。必须达到分子学转阴后方可开始维持治疗。
每3个月为1个周期,第1个月:ATRA 25 mg·m-2·d-1×14 d,间歇14 d;第2个月和第3个月:亚砷酸0.16 mg·m-2·d-1或复方黄黛片60 mg·m-2·d-1×14 d,间歇14 d。完成8个周期,维持治疗期总计约2年。
ATRA 25 mg·m-2·d-1直到CR,DNR 45 mg·m-2·d-1静脉注射或IDA 8 mg·m-2·d-1静脉注射,第2、4、6天。
ATRA 25 mg·m-2·d-1×14 d+ DNR(45 mg·m-2·d-1静脉注射)或IDA(8 mg·m-2·d-1静脉注射)×3 d,间歇28 d,为1个疗程。共2个疗程。
每3个月为1个周期:ATRA 25 mg·m-2·d-1,第1~14天;6-巯基嘌呤(6-MP)50~90 mg·m-2·d-1,第15~90天;甲氨蝶呤(MTX)5~15 mg/m2,每周1次,共11次。共8个周期,维持治疗期总计约2年余。
ATRA 25 mg·m-2·d-1联合亚砷酸0.16 mg·kg-1·d-1或复方黄黛片60 mg·kg-1·d-1,直到CR;DNR 45 mg·m-2·d-1或IDA 8 mg·m-2·d-1第1~3天。
可选用以下方案:
①HA方案:HHT 2 mg·m-2·d-1,第1~7天;Ara-C 100 mg·m-2·d-1,第1~5天。
②MA方案:MIT 6~8 mg·m-2·d-1,第1~3天;Ara-C 100 mg·m-2·d-1,第1~5天。
③DA方案:DNR 45 mg·m-2·d-1,第1~3天;Ara-C 100 mg·m-2·d-1,第1~5天。
④IA方案:IDA 8 mg·m-2·d-1,第1~3天;Ara-C 100 mg·m-2·d-1,第1~5天。
若第3次巩固化疗后未达到分子学转阴,可加用IDA(8 mg·m-2·d-1,第1~3天)和Ara-C (1.0 g/m2,每12 h 1次,第1~3天)。必须达到分子学转阴后方可开始维持治疗。
每3个月为1个周期,第1个月:ATRA 25 mg·m-2·d-1×14 d,间歇14 d;第2个月和第3个月:亚砷酸0.16 mg·kg-1·d-1或复方黄黛片60 mg·kg-1·d-1×14 d,间歇14 d。完成8个周期,维持治疗期总计约2年。
ATRA 25 mg·m-2·d-1,第1~36天;亚砷酸0.16 mg·kg-1·d-1,第9~36天;IDA 6~12 mg·m-2·d-1,静脉注射,第2、4、6、8天。
①ATRA 25 mg·m-2·d-1,第1~28天+亚砷酸0.16 mg·kg-1·d-1,第1~28天;②ATRA 25 mg·m-2·d-1,第1~7、15~21、29~35天+亚砷酸0.16 mg·kg-1·d-1,第1~5、8~12、15~19、22~26、29~33天。
每3个月为1个周期:ATRA 25 mg·m-2·d-1,第1~14天;6-MP 50~90 mg·m-2·d-1,第15~90天;MTX 5~15 mg/m2,每周1次,共11次。共8个周期,维持治疗期总计约2年余。
ATRA的诱导分化作用可以持续较长时间,在诱导治疗后较早行骨髓评价可能不能反映实际情况。因此,骨髓形态学评价一般在第4~6周、血细胞计数恢复后进行,此时,细胞遗传学一般正常,而PML-RARα或发病时相应异常基因转录本在多数患者仍为阳性。CR标准同其他AML[16,17]。
建议采用定量PCR监测骨髓PML-RARα转录本水平[18],治疗期间建议2~3个月进行1次分子学反应评估,持续监测2年。上述融合基因持续阴性者继续维持治疗,融合基因阳性者4周内复查。复查阴性者继续维持治疗,确实阳性者按复发处理。流式细胞术因对于APL的MRD敏感性显著小于定量PCR,因此不建议单纯采用流式细胞术对APL进行MRD监测。
五、支持及其他治疗
1.临床凝血功能障碍和出血症状严重者:首选为原发病的治疗。支持治疗如下:输注单采血小板以维持PLT≥(30~50)×109/L;输注冷沉淀、纤维蛋白原、凝血酶原复合物和冰冻血浆维持纤维蛋白原>1500 mg/L及PT和APTT值接近正常。每日监测DIC相关指标直至凝血功能正常。如有纤溶异常,应快速给予ATRA。如有器官大出血,可应用重组人凝血因子Ⅶa。
2.高白细胞APL患者的治疗:不推荐白细胞分离术。可给予水化及化疗药物。
3.APL分化综合征[19]:临床表现为以下7个表现:不明原因发热、呼吸困难、胸腔或心包积液、肺部浸润、肾脏衰竭、低血压、体重增加5 kg,符合2~3个者属于轻度分化综合征,符合4个或更多个者属于重度分化综合征。分化综合征通常发生于初诊或复发患者,WBC>10×109/L并持续增长者,应考虑停用ATRA或亚砷酸,或者减量,并密切关注体液容量负荷和肺功能状态,尽早使用地塞米松(10 mg,静脉注射,每日2次)直至低氧血症解除。
4.砷剂不良反应监测:治疗前进行心电图(评估有无QT间期延长)检查,外周血的肝功能和肾功能相关检查;同时要注意口服砷剂患者的消化道反应。
5.CNSL的预防和治疗:低中危APL患者,ATRA联合砷剂作为一线治疗方案中建议预防性鞘内治疗;高危APL或复发患者,因发生CNSL的风险增加,对这些患者应进行至少2~6次预防性鞘内治疗。对于已诊断CNSL患者,按照CNSL常规鞘内方案执行。
6.APL诱导治疗期间不主张应用G-CSF。
7.对于有高凝及血栓形成的患者可应用抗凝药物进行治疗。
8.肺功能损害:治疗中应注意肺功能情况。
9.肾功能损害:间断复查肾功能,防止肾功能损害的出现。
(执笔:马军)
参加指南讨论的专家:中国医学科学院血液学研究所、血液病医院(王建祥、秘营昌);上海交通大学附属瑞金医院(沈志祥、李军民、赵维莅);北京大学人民医院、北京大学血液病研究所(黄晓军、江滨、主鸿鹄);哈尔滨血液病肿瘤研究所(马军、邱林);苏州大学附属第一医院(吴德沛);浙江大学附属第一医院(金洁);四川大学附属华西医院(刘霆);福建医科大学附属协和医院(胡建达)
更正:
发表于本刊2018年第3期“中国急性早幼粒细胞白血病诊疗指南(2018年版)”一文第180页(二)预后分层
1. ATRA联合化疗作为一线治疗模式下的预后分层:
(1)低危:WBC <10×109/L,PLT ≥40×109/L。
(2)中危:WBC <10×109/L,PLT <40×109/L。
(3)高危:WBC ≥10×109/L。
2. ATRA联合砷剂作为一线治疗模式下的预后分层:
(1)低危:WBC <10×109/L。
(2)高危:WBC ≥10×109/L。
更正为:
1. ATRA联合化疗作为一线治疗模式下的预后分层:
(1)低危:WBC ≤10×109/L,PLT ≥40×109/L。
(2)中危:WBC ≤10×109/L,PLT <40×109/L。
(3)高危:WBC >10×109/L。
2. ATRA联合砷剂作为一线治疗模式下的预后分层:
(1)低危:WBC ≤10×109/L。
(2)高危:WBC >10×109/L。
[19]. Differentiation syndrome in patients with acute promyelocytic leukemia treated with all-trans retinoic acid and anthracycline chemotherapy: characteristics, outcome, and prognostic factors[J]. Blood, 2009,113(4):775-783. DOI:10.1182/blood-2008-07-168617.