中国抗癌协会
立即下载App多参数流式细胞术检测急性白血病及浆细胞肿瘤微小残留病中国专家共识(2017年版)
多参数流式细胞术(multiparameter flow cytometry, MP-FCM)是运用不同荧光标记的多种抗体组合对造血细胞表面或胞内抗原的表达状况进行检测,进而对细胞的系列来源、分化程度、表型异常与否进行分析判断的高通量、高敏感性检测技术,其在恶性血液病的诊断分型、治疗监测、预后评估及治疗靶点筛查中已成为必不可少的实验诊断手段[1,2]。微小残留病(minimal residual disease, MRD)是指恶性血液病经过治疗达到血液学完全缓解后体内残存的通过形态学等传统方法无法检测到的任何水平的微量肿瘤细胞状态[3,4]。近年来,也有学者根据现有检测技术的局限性将MRD定义为可检测的残留病(measurable residual disease)。运用FCM检测恶性血液病MRD的研究始于20世纪80年代,利用2~4色FCM对急性白血病的治疗后样本进行检测,并基于在正常细胞分布之外的"空白"区域检测到异常表型的理论对MRD进行分析鉴别[5,6];在过去的10年中,随着FCM水平的不断提高,特别是8~10色MP-FCM在MRD检测中体现出敏感性高、特异性好、适用范围广的优势,MRD的FCM检测从小规模样本的临床研究发展到大规模样本的临床验证,目前已成为恶性血液病临床疗效判断和预后分层的重要依据,而基于MRD指导下的个体化治疗可以使不同危险度分层组中的患者避免过度治疗或治疗不足,以提高患者的长期生存率[7,8,9]。
然而,由于白血病等恶性血液病存在不同程度的克隆异质性、不同治疗方案对免疫表型和克隆筛选的影响、化疗后不同时间点骨髓造血恢复的状况不一、不同基因背景的残留肿瘤细胞在复发时的增殖动力学不同等疾病自身因素的复杂性,以及不同检测中心在抗体组合、荧光搭配、样本处理、圈门策略、分析逻辑、主观经验、质控管理等诸多技术环节中的差异,目前国内外对于MRD的检测和分析还缺乏规范化,导致对MRD检测结果的临床解读和预后评估标准存在不一致性。为了使国内各检测中心对于MRD的FCM检测和分析逐步实现规范化和标准化,在可控因素范围内尽量减少检测结果之间的差异,中国免疫学会血液免疫分会临床流式细胞术学组通过各种学术讨论、多中心室间比对等一系列努力,形成了基于MP-FCM检测MRD的国内共识,供大家参考,也希望在此基础上不断完善,以期提高我国恶性血液病MRD检测的整体水平。本共识涉及病种包括急性髓系白血病(aute myeloid leukemia, AML)、急性T淋巴细胞白血病(T-cell precursor acute lymphoblastic leukemia/lymphoma, T-ALL),急性B淋巴细胞白血病(B-cell precursor acute lymphoblastic leukemia/lymphoma, B-ALL)和浆细胞肿瘤(plasma cell neoplasms, PCN)。
FCM检测MRD的原理是通过分析细胞表面或胞内一系列抗原的表达模式来识别只在白血病细胞中出现而正常骨髓中不存在或存在比例极低的免疫表型,即"白血病相关免疫表型"(leukemia-associated immunophenotypes, LAIP)。根据抗原表达特点将LAIP分为以下几种表现形式:跨系抗原表达、非同步抗原表达、抗原表达量异常、散射光信号异常以及表达少数白血病特异性抗原(如NG2)。跨系抗原是指某一系列的细胞表达其他系列的抗原,如B-ALL的白血病细胞中表达髓系抗原CD66C、CD13、CD33等;非同步抗原表达是指应该分别出现在分化早期和晚期的抗原同时出现,如AML中CD11b(晚期)和CD34(早期)共表达;抗原表达量异常指与同系列、相同分化阶段的正常造血细胞比较,白血病细胞中某些抗原的表达强度明显异常,包括减低(low expression,low)、缺失或升高(bright expression,bri),如正常CD19+CD34+B系原始淋巴细胞应该高表达CD38,B-ALL中白血病细胞则低表达甚至不表达CD38。另外,白血病细胞经常出现前向散射光(forward scatter, FSC)和侧向散射光(side scatter, SSC)的改变,也可以作为识别白血病细胞的依据[10,11]。
检测MRD选用的抗原主要包括三部分:①设门所用的抗原,为系列敏感性高的抗原,能初步圈定某一系列所有细胞,如T系的胞质CD3(cyCD3)或CD7、B系的CD19、髓系的CD117,以及浆细胞的CD38和CD138。②确定相应系列早期分化的抗原,可识别该系列原始、幼稚细胞群体及亚型,如B系的CD45、CD10、CD34和核TdT(nTdT),T系的膜CD3(mCD3)、CD34、nTdT和CD99,髓系的CD34、CD117、HLA-DR和CD38。③白血病相关的异常抗原,能区分白血病细胞和对应系列的正常幼稚细胞,主要指LAIP涉及的抗原。前两部分抗原虽然有上述的作用,但同时这些抗原在白血病中也会出现抗原表达的异常,也可以表现为LAIP,如B-ALL中CD10强表达、CD20和CD34共表达等均可以作为跟踪白血病细胞的依据,因此具有多方面的作用[12]。
在MRD数据分析的实际应用中,主要存在2种方法:第一种,根据患者发病时的LAIP设计特异性的抗体组合,选择固定的门(gate)对特定的细胞群进行设门分析,例如对发病时具有CD10+CD38- LAIP特征的B-ALL患者,通常以CD19+细胞设门,重点分析是否存在CD10+CD38-细胞;第二种,始终以同系列相同分化阶段的正常细胞作为对照,在每次检测时观察是否存在与正常细胞表型不同的细胞群体,即"不同于正常细胞表型"(different-from-normal,DFN)的方法。
第一种方法的优点是对于初学者比较容易掌握,但缺点是依赖初治时的免疫表型,如果初治时没有检测免疫分型,则无法进行MRD检测;另外,在疾病的进展过程中,由于不同治疗方案对白血病细胞分化、克隆筛选以及表型漂移等因素的影响,仅依据初治LAIP及固定门分析MRD时,有可能导致假阴性结果[9,13,14,15]。第二种方法避免了第一种方法的缺点,在MRD监测过程中可以及时发现病情变化后新出现的LAIP表型,提高检测的敏感性;但需要检测者熟练掌握正常细胞不同系列、不同分化阶段、不同种抗原表达的规律,包括抗原出现的时序性及抗原表达量的强弱变化规律。因此,对检查者的经验要求较高。而对于AML患者,因抗原变化较复杂,为避免漏诊,需要每次筛查较多的抗原,从而会增加每次检测的费用。对于T-ALL及B-ALL,目前研究表明,单管8~10种抗原同时检测,几乎可以覆盖所有的患者。我们主张两种方法要有机地结合起来,即如果能够依据第一种方法的原则进行检测时,也要兼顾第二种方法的原则,既要关注发病时的LAIP,同时也要关注是否出现新的"DFN"的LAIP,以发挥第一种检测方法的优点及克服其缺点,达到既经济又准确的结果。但如果不具备初治时免疫表型,只能采用第二种方法进行检测。
基于MRD检测的方法不同,MRD检测时抗体组合的选择主要参照以下原则:①有初发LAIP可供参考时,尽可能多地将LAIP涉及的抗原包括在MRD检测的组合中。②对于没有初发时免疫表型的患者,治疗后如果需要检测MRD,只能参考同类型的白血病出现LAIP的频率,选择出现频率高的抗原及选取较多的抗体进行检测,以免漏检。
为了简化MRD抗体组合的复杂性,本学组专家以两种方法中的相关抗原以及高频出现异常表达抗原的抗体作为骨架抗体,提出了不同类型白血病进行MRD检测较通用的基本抗体组合,在此基础上,再根据不同患者的免疫表型个体化地补充第三类抗体(备选抗体),成为较完整的MRD抗体组合,进行临床检测。
B-ALL MRD检测的关键是要与正常幼稚B淋巴细胞进行鉴别,尤其是在婴幼儿、青少年及化疗或造血干细胞移植后,骨髓中会出现大量再生的幼稚B淋巴细胞时。B淋巴细胞系列和分化阶段相关抗原的抗体如CD19、CD10、CD34、CD20和CD45首选为"骨架"抗体,同时CD10、CD19和CD34的强表达,CD45低表达以及CD20和CD34共表达等特征也可以作为识别白血病细胞的依据;其次入选在B-ALL中高频出现异常表达抗原的抗体,如CD38(约60%)、CD81(约80%)和CD58(约50%),其中CD38和CD81在B-ALL中常见低表达甚至阴性,而CD58则表现为过表达且治疗过程中表达较稳定;最后根据患者的免疫表型特征个体化地选择其他的LAIP的标志,如CD13、CD33、CD66C、CD15、CD65、CD73、CD123、CD304和CD86等,以及所谓的白血病特异性抗原如NG2(anti-7.1)。其中NG2的表达与MLL基因重排高度相关,该组基因重排患者除了特征性表达NG2外,主要免疫表型特征为CD19+CD34+/-nTdT+CD10-CD15+或CD65+。BCR-ABL阳性患者则高频表达髓系抗原如CD66C、CD13或CD33,同时CD10+CD34briCD38low。另外,nTdT+CD34-、CD21+CD34+、cyIgM(cμ)+CD34+、cμ+nTdT+等非同步抗原表达也可作为跟踪白血病细胞的依据。值得注意的是nTdT、CD34和CD10等B系早期分化抗原在诱导治疗阶段易出现抗原表达下调现象,而晚期分化抗原如CD20则常表达上调,这可能与皮质激素的诱导细胞分化作用相关[12, 16,17,18]。另外,随着嵌合抗原受体(CAR)-T细胞治疗的应用,CD19在白血病细胞上的表达可能会减弱或缺失,因此在CAR-T-CD19治疗后,仅用CD19阳性细胞设门来圈出B淋巴细胞群体容易导致假阴性结果;有研究者建议增加CD22和(或)CD24设门可以避免这一现象[19]。同理,部分CD20阳性的B-ALL患者如选择CD20单克隆抗体治疗,MRD检测时不应仅关注CD20阳性异常B淋巴细胞。
胸腺是正常T淋巴细胞分化发育的主要器官,骨髓和外周血中几乎检测不到幼稚T淋巴细胞,因此T-ALL的MRD检测基本不存在正常幼稚T淋巴细胞背景的干扰。在T-ALL MRD检测中,判断骨髓或外周血中是否存在异常幼稚T淋巴细胞是检测的关键。根据T淋巴细胞在胸腺中的抗原表达规律,幼稚T淋巴细胞相关的标志包括:CD34、nTdT、CD99、CD1a、CD10、cyCD3+/mCD3-、mCD3-CD4+CD8+等。文献报道约90%的T-ALL患者表达CD99或nTdT,40%表达CD34,但这些抗原在治疗早期经常出现表达下调。但是,这些抗原仍是鉴定幼稚T淋巴细胞较好的标志。大部分T-ALL患者不表达或低表达mCD3,而cyCD3阳性,CD7多见异常高表达,因此最好采用cyCD3进行设门,"骨架"抗体中应该包含mCD3、cyCD3、CD7、CD45。可以依据mCD3、cyCD3和CD45的表达判断是否存在幼稚T淋巴细胞。在T-ALL中,CD5低表达多见,尤其在ETP(early T precursor acute lymphocytic leukemia)中。CD2在pro-T-ALL中表达缺失,组合中最好同时包含这些抗原;由于CD2和CD7在NK和T淋巴细胞中都可以表达,而且部分NK细胞也可以表达cyCD3,因此MRD检测时最好加入CD56和(或)CD16以去除cyCD3+mCD3-NK细胞群体的干扰。如果T-ALL初发时表达CD56,则也可将CD56作为异常抗原。其他在T-ALL中表达而正常骨髓或外周血T淋巴细胞一般不表达的抗原,如CD13、CD33、CD117、CD11b和CD65等,则可以根据具体情况选用相同荧光素标记。CD44在T-ALL中常可见异常低表达,也可在MRD检测组合中使用。CD4和CD8在T-ALL中可以出现双阴性、双阳性或单阳性,在荧光通道有空余或者通过上述标志不能鉴别异常T淋巴细胞时,可选择这两种抗原。文献报道CD3、CD4、CD8、CD2、CD5、CD7治疗后表达较稳定[14,20],但要注意,只出现CD3、CD2、CD5、CD7表达强度异常,甚至CD4+CD8+,也可见于周围型淋巴瘤,只有结合初治时表型,或者同时具有其他幼稚T淋巴细胞的表型才能判断为MRD阳性。
髓系原始细胞通常表现为CD34+CD117+或者CD34-CD117+,HLA-DR在髓系不同阶段和亚型中表达有所不同。因此,CD34、CD117、HLA-DR、CD45入选为AML的"骨架"抗体。另外CD13和CD33的表达情况可以较好地区分正常和异常造血,CD38在正常原始细胞中高表达,而白血病细胞中常见低表达,尤其是骨髓增生异常综合征转化的AML[7]。除上述抗原以外,交叉抗原对于区分正常和异常造血很重要,也应该入选,如CD56、CD19、CD2、CD4和CD5等。多个交叉抗原同时表达时可以选用同种荧光素标记。由于CD7在AML中表达较为特殊(正常髓系原始细胞中可见低表达),建议单独标记。同时,初发时若存在非同步抗原表达如CD34+CD11b+、CD34+CD64+、CD34+CD15+等,MRD检测组合中也可将这些抗原入选。CD123在AML中可见表达明显上调,NG2的异常表达在AML中也有一定的发生频率,初发时如果阳性,这两种抗原也可入选。因为骨髓中CD34+细胞群体主要包含CD10+B系原始细胞和CD10-髓系原始细胞,如果检测通道有空余的话,建议AML的MRD检测组合中加入CD10,尤其是CD34+CD117low/-AML,去除CD10+CD34+B系原始细胞群体更有利于识别异常髓系原始细胞;同时也可以观察骨髓B系的重建状态[21]。对于急性早幼粒细胞白血病(APL)而言,PML-RARα融合基因检测的特异性和灵敏度都显著高于FCM,在此不做特别的抗体推荐。
CD38和CD138通常用来界定浆细胞群体,CD19和CD56可以较好地区分正常和异常浆细胞,PCN的"骨架"抗体应包含CD38、CD138、CD56、CD19、CD45。正常浆细胞群体CD38bri CD138bri且非限制性表达胞质免疫球蛋白kappa或lambda轻链(cyκ或cyλ)。绝大部分正常浆细胞为CD19+CD56-CD45+CD20-CD27+CD28-CD81+CD117-CD200-/+,符合典型的正常浆细胞免疫表型特征;有一小部分正常浆细胞(低于总浆细胞群体的30%)可以CD19-或CD56+或CD45-/low或CD20+或CD27low或CD28+或CD200bri,这些非典型的免疫表型一般不会同时出现,最重要的是cyκ/cyλ为非限制性表达。异常浆细胞的免疫表型可以表现为CD38、CD19、CD45、CD27、CD81的异常低表达或不表达,以及CD20、CD28、CD56、CD117、CD200的异常表达,而且限制性表达cyκ或cyλ轻链。CD81low、CD117+和CD38low一般只出现在异常浆细胞中,特异性较高,建议MRD检测组合中包含这些抗原。CD27在大部分正常浆细胞中高表达,而PCN中可见表达明显减低,而且发生频率较高,也可包含在检测组合中[22,23]。由于检测胞质免疫球蛋白轻链(cyκ和cyλ)需要洗涤、透膜,步骤较多,易造成细胞的丢失,因此国外的有些学者认为在膜抗体检测数量足够多的情况下(这些组合中往往包括CD27、CD81等8种抗体)可以不用检测cyκ和cyλ。关于是否需要检测cyκ和cyλ,目前国内外仍存在争议。本学组专家认为胞质免疫球蛋白轻链的限制性表达对于确定克隆性浆细胞是最直接的证据,在使用抗体相对较少时,还是建议进行检测,因此这两种抗原也纳入骨架抗体中。异常浆细胞还可以表达CD13、CD33等交叉抗原,其他抗原如CD28在PCN中表达上调,可视仪器配置情况酌情选用。值得注意的是,部分骨髓瘤患者的异常浆细胞可以低表达或者不表达CD138,初步设门圈定浆细胞群体时要注意结合其他标志。
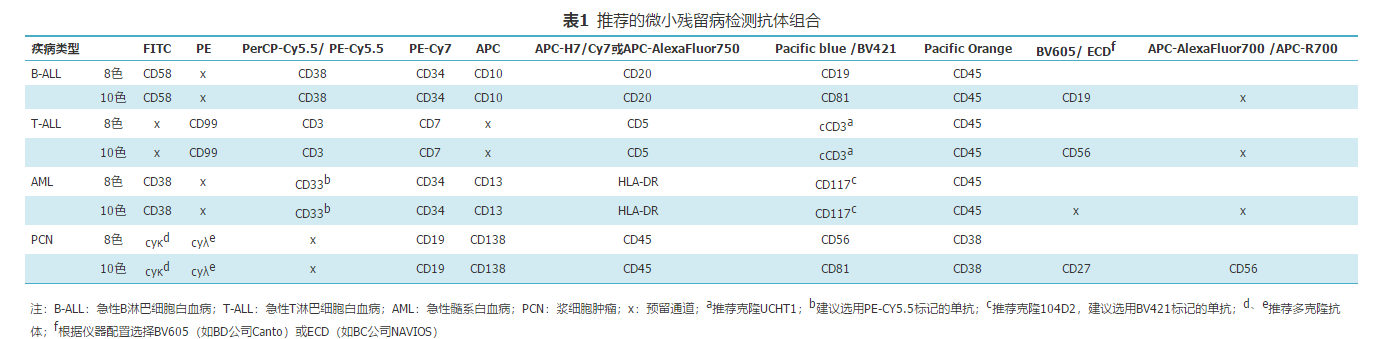
表1 推荐的微小残留病检测抗体组合
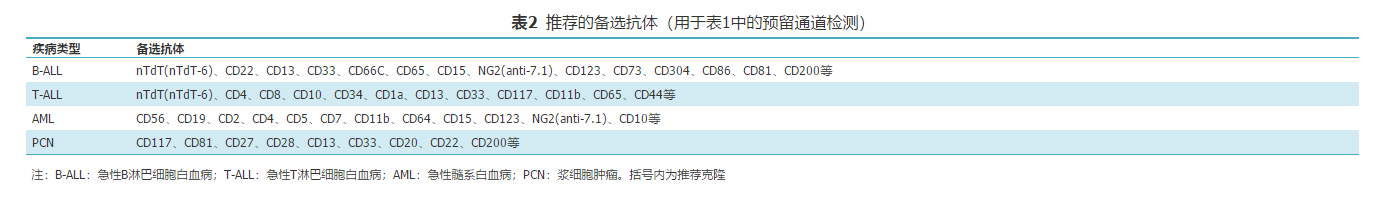
表2 推荐的备选抗体(用于表1中的预留通道检测)
有多种因素影响患者的预后分层,如患者的年龄、发病时白细胞计数、白血病亚型、肿瘤细胞的遗传学背景等,这些主要是治疗前相关预后因素。而白血病MRD是一个治疗后相关的预后分层因素,反映的是肿瘤细胞对不同治疗方案的敏感性,因此MRD的检测和结果解读与治疗过程中诸多环节密切相关。各检测中心应与临床进行较好的沟通与衔接,才能在检测样本的选择、检测时间点和检测频率的确定、检测阈值的界定、不同治疗方案对于骨髓造血恢复的影响、挽救治疗的时机选择等方面达到较好共识,进而对MRD检测结果的解释和判断更加客观准确,真正实现MRD指导下的个体化治疗。
由于不同类型恶性血液病的细胞起源、遗传学背景、生物学特征、药物敏感性等因素的影响,化疗后不同时期,骨髓和外周血中肿瘤细胞的清除速率存在不同程度的差异。
在AML的研究中发现,在化疗后早期(3~7 d),由于尚未达到形态学缓解,外周血中还存在一定负荷的肿瘤细胞,因此在早期采取创伤性较小的外周血检测,背景细胞的干扰很少,MRD检测的敏感性和特异性较高。通过计算肿瘤负荷下降的速度可以判断其对治疗方案的敏感性,对于疾病的预后分层有一定的指导意义。而在达到形态学缓解后,骨髓的检测要明显优于外周血检测,二者的检测敏感性至少相差一个对数级[24,25,26]。
国外的大样本研究证实,T-ALL的MRD检测由于具有比较特异的LAIP,因此较易在骨髓和外周血中检出,T-ALL的MRD检测在外周血的检测敏感性大致接近或略低于骨髓(约减低1个对数级),因此在治疗早期骨髓和外周血的检测敏感性接近[8, 27,28]。
在B-ALL的MRD检测对比研究中发现,外周血的MRD检测敏感性比同期骨髓减低了1~3个对数级,因此对于B-ALL的MRD检测建议首选骨髓。另外,由于儿童B-ALL治疗方案已实现标准化,并且方案中已纳入了MRD的指导策略,国外对于骨髓样本的抽取强烈推荐选取第一管骨髓,以避免在抽取过程中造成不同程度的外周血稀释,而且建议抽取量不宜过多(一般在2 ml左右,<5 ml)。当然,样本的留取量要保证检测的敏感性,如要达到10-5的检测敏感性,并以检出≥50个异常细胞为判断标准,则抽取细胞数需≥1×107[8,27,28]。
化疗后不同时间点的MRD检测对于预后的指导意义不同,大致可分为早期和晚期两个阶段。第一阶段为诱导治疗后MRD监测,用于指导早期危险度分层和下一步巩固治疗策略的选择;第二阶段为巩固治疗后MRD监测,用于指导维持治疗的强度以及出现MRD水平复发时的挽救治疗策略。对于AML而言,在诱导治疗后、巩固治疗后、移植前以及移植后的任何时间点的MRD阳性都有不同程度的预后价值,一旦出现阳性即意味着治疗不是完全有效并且预示复发的风险增大[29,30,31]。而对于ALL而言,化疗后早期(3个月内)MRD阴性提示预后好,早期MRD≥10-2或10-3提示预后不良;在化疗后期(巩固治疗后、再诱导后及维持治疗的前期)任何水平的MRD阳性都提示预后不良。大宗样本研究显示,儿童ALL的MRD监测在最初3个月对于分层的指导意义更大[32,33,34]。
化疗后骨髓造血细胞的恢复存在明显的时间相关性,化疗后第15~33天白细胞计数较低,而化疗后第79天白细胞计数较高。因此,化疗后第15天检测骨髓MRD时,因为肿瘤负荷还相对较高,MRD阳性检出率受白细胞计数的影响较小;而化疗后第33天检测MRD时,可能由于骨髓中肿瘤细胞和白细胞数量均较低,MRD检测敏感性往往≤10-4,因此获取足够的细胞数对于提高检测敏感度很重要[8]。在诱导治疗后或移植治疗前期,骨髓中较早恢复的是幼稚髓系细胞,因此对于AML而言,MRD检测的背景干扰较大,而B-ALL的检测干扰较小;但在巩固治疗后或移植治疗后期,骨髓中的幼稚细胞以不成熟B淋巴细胞为主,因此B-ALL的MRD检测背景干扰增大,而AML的MRD检测的干扰相对较小[7,35]。
由于白血病的遗传学背景、生物学特性不同,导致肿瘤细胞的增殖动力学不一致,因此不同类型的白血病从分子水平复发到血液学复发所需的时间也不尽相同。所以,MRD检测频率的确定很难界定统一标准。德国多中心ALL(German Multicenter ALL, GMALL)临床试验结果表明,成人Ph染色体阳性B-ALL从MRD转阳到血液学复发的中位时间为4.1个月[36]。而在AML的研究中发现,携带RUNX1-RUNX1T1融合基因的白血病细胞的平均倍增时间为14 d,携带CBFβ-MYH11融合基因的白血病细胞的平均倍增时间为36 d,合并FLT3-ITD突变和NPM1突变的细胞比伴单纯NPM1突变的细胞的平均倍增时间降低了近50%(7.4 d对15 d)[37]。因此,建议根据疾病的不同类型和遗传学特征制定个体化的检测频率。另外,在不同病种、不同年龄和不同治疗方案下,疾病缓解的持续时间是不同的。例如儿童B-ALL的疗效较好,缓解持续时间一般较长,只有35%左右的患儿在2年内复发,因此MRD的监测频率可适当减少;而对于儿童T-ALL,约95%的患儿在整个治疗过程中都有可能出现复发,因此需要适当增加MRD的监测频率;成人ALL的缓解持续时间较短,因此增加MRD的监测频率有助于预测早期复发[8]。总之,对于复发危险度较高的患者,检测频率要高于复发危险度低的患者。
目前,关于MRD阈值的研究报道大多是基于MRD检测值与临床预后的相关性来确定的。在AML的临床研究中发现,MRD>3.5×10-4或1.0×10-3提示高复发风险和预后不良;而关于ALL的研究显示,MRD>1.0×10-4或1.0×10-3则提示预后不良。由于在不同研究中,患者入组标准及治疗方案不同、MRD检测所依据的异常表型的敏感性及特异性不同,因此不同临床研究结果中的MRD阈值不具有可比性,也难于推广[7,8,9]。
从检测的技术角度分析,MRD的阳性阈值即阴、阳性界限(cutoff值)的界定取决于以下几个主要因素:患者自身白血病细胞的LAIP的特异性和敏感度、治疗后不同时间点的样本中正常细胞背景的干扰程度、不同治疗方案对白血病克隆选择性清除和表型漂移的影响等。只有在检测技术上不断规范和精准界定MRD的cutoff值,才能使MRD阈值与临床预后具有更好的相关性和可比性。
MRD的检测敏感性通常由以下三个因素决定:①可重复地定量鉴定MRD阳性细胞群体的最小细胞数:这个阈值指的是最低定量限(lower limit of quantification, LLOQ),而不是最低检出限(limit of detection, LOD)。LLOQ=(可重复地定量鉴定MRD阳性群体的最小细胞数/总细胞数)×100%。国际上的研究认为20个细胞是LOD的比较保守的值,考虑到计数误差,以95%可信限上限作为标准,LOD=30/获取细胞数×100%。而50个细胞是普遍接受的可重复定量检测的最低细胞数,因此LLOQ=50/获取细胞数×100%。如果获取500 000个细胞,则LOD和LLOQ分别为0.006%和0.01%[22, 38]。②LAIP的特异性:指区分正常与异常造血的能力,所选LAIP在正常骨髓中的本底水平越低,则特异性越好[7]。③获取的总细胞数:特异性足够好的话,理论上获取的细胞总数越高,敏感性就越高。文献报道获取细胞总数达5×106时,ALL和PCN MRD检测的敏感性均可达10-5。对于AML,由于多数患者的LAIP特异性较低,目前普遍认为敏感性只能达到10-3,但是如果LAIP的特异性较高时,有一部分AML患者的敏感性可以达到10-4,部分患者甚至可以达到10-5。
LAIP特异性是影响MRD阈值界定的关键因素[7,8,9]。依据特异性不同,可将LAIP分为2种类型:一种是在正常骨髓中完全不存在而只在白血病细胞上存在的表型,例如抗原表达增强、减弱或缺失,这类LAIP与正常细胞发育轨迹完全没有重叠,因而具有较高的特异性和敏感性。第二种LAIP是在正常骨髓中存在较低水平的表达,例如AML中存在髓系幼稚细胞低水平表达CD7或CD2、CD34-CD117+CD11b+表型、CD34+CD33+CD56+表型等,这类表型在化疗后的骨髓恢复期或者外源性生长因子治疗后的正常髓系幼稚细胞上均有可能出现,因而导致其背景值(或噪音)增高,特异性和敏感性降低;而且,不同的LAIP在正常骨髓中表达的背景值也是不同的,因此,需要根据它们在正常骨髓幼稚细胞中表达的中位值来分别确定MRD检测的cutoff值。上述两种LAIP的cutoff值与检测敏感度之间的相关性是完全不同的。对于前者,只要把检测质控做好,一旦出现具有LAIP的细胞,在排除非特异性染色后即可认为是白血病细胞,其检出的敏感性与获取的细胞数完全成正比,只要获取足够多的细胞,其敏感性甚至可以达到10-6,接近或达到定量PCR水平。而对于第二类LAIP,因为其存在一定的背景值,即使增加获取的细胞数,也不能进一步提高敏感性,只有当检测值高于背景值时才能确定为MRD阳性,这给MRD检测的标准化带来了极大难度。因为不同仪器的参数设定、不同的荧光搭配及抗体克隆号的选择均有可能导致同一类型的LAIP在不同实验室之间背景值(即检测cutoff值)不同。针对这一类LAIP,国外的学者采取比较治疗前后白血病细胞减少的对数值(Log difference, LD)的方法,而不是直接比较LAIP+细胞比例的变化[39]。
四、MRD检测的质量控制与标准化
除去前面提到的多种因素外,FCM的质控也至关重要。如果质控做得不好,直接影响结果的准确性和特异性,因此将质控也纳入本共识中。本学组同期发布的《白血病/淋巴瘤免疫分型检测质量控制指南》将从分析前、分析中和分析后各检测环节中对检测质量有影响的各技术要点,为临床医师、护理人员、样本运送人员、检测人员和检测报告审核人员提供可操作性强的实施细则,其中任何一个环节的失控都可能导致结果的偏差。本共识仅根据MRD检测的特殊性提出以下注意事项[9, 12, 38, 40],具体的细则还需参照《白血病/淋巴瘤免疫分型检测质量控制指南》。
(1)光路校准:每天1次(一般为开机后)利用微球校准散射光和荧光(强度和CV值)。对于拥有多台流式细胞仪的实验室,不同仪器之间(包括同一型号)应进行平行比较,每6个月1次。用不同的仪器检测同一份样本(MRD水平接近LOD),所测得的结果CV值不应超过30%。
(2)荧光补偿校准:荧光补偿与流式细胞仪光电倍增管的电压、增益,激光功率,光学滤片等有关,这些参数有变动时必须马上调整补偿值。
(3)预防交叉污染:检测交叉污染(carryover)的方法:依次检测以下样本:MRD接近LOD(≈0.01%)→较高水平MRD→MRD接近LOD,观察前后两次MRD低水平标本结果的一致性;两管之间的交叉污染不能超过检测特异性;交叉污染严重时,应在每个测试管之间插入装有清洁水(三蒸水、去离子水等)的管子进行冲洗,自动上样或孔板上样时尤其要注意。
(1)精确度/重复性:将已知数量的异常细胞混入正常骨髓标本中,3复管进行检测,计算CV%,最好<10%。当MRD水平很低(如接近LOD)时,<30%也可以接受。
(2)特异性和敏感性:详见第二、第三部分。
(3)稳定性:①样本稳定性:取MRD水平接近LOD的样本,每隔6 h监测1次,观察结果的稳定性,以确定样本的稳定性范围。收到标本后应尽快进行MRD检测,对于无法及时进行MRD检测的实验室(EDTA抗凝样本>24 h或者肝素钠抗凝样本>48 h),必须要保证样本在稳定性范围内进行检测。如果样本保存时间>24 h,需进行细胞活力评估,活力<85%或90%时,不予报告结果。不可替代样本(如骨髓)除外,但需提示标本活力。活力欠佳的标本,非特异性结合高,某些抗原表达下调(如CD138),某些脆弱细胞群(如浆细胞群体)很可能会丢失。标本有凝块时应在报告中注明。②验证混合抗体使用中的稳定性:正式使用自行配置的多色预混抗体前,应每周1~2次,取接近LOD的样本,同时用提前配置的混合抗体和新鲜混合的抗体进行标记,比较结果的一致性,以确认混合抗体的稳定效期。确定效期后,建议每次根据实际标本量配置混合抗体的份数,尽量在有效期内用完。
(4)数据收集:①建议每管中加入待标记的白细胞总数为2×106,上机获取1×106个白细胞。对于MRD检测敏感性要求更高的实验室,如预期敏感性为1×10-5时,则获取细胞总数需达2×106。②本共识推荐使用全血或全骨髓溶血标记法。根据预计待标记细胞数量确定所需标本体积:所需标本体积≤100 μl时,染色后进行红细胞裂解;标本体积>100 μl时,先裂解红细胞(注意选用不含固定成分的红细胞裂解液,例如氯化铵裂解液),离心富集于100 μl体系中,方可进行混合抗体染色。具体的抗体组合优化及染色步骤可参照《白血病/淋巴瘤免疫分型检测质量控制指南》。
1.在开展MRD检测项目前,必须利用相同的抗体组合至少分析20份正常对照标本的抗原表达模式,建立正常骨髓发育模板。正常对照标本应包含正常和重建(化疗后或移植后)骨髓。同时计算每种LAIP在正常骨髓中的本底水平(背景值),MRD检测时对于某种LAIP而言,只有比例高于该水平才能认为MRD阳性。
2.对于有初发免疫分型结果的患者,首选初发时筛选出的异常抗原(尽可能同时监测多种LAIP),对治疗后的骨髓标本进行分析。注意分析是否存在发病时LAIP+细胞,同时利用"DFN"的方法观察是否存在发生免疫表型变异或克隆转化的异常细胞群体。
3.如果发病时没有进行免疫分型检测,治疗后每次进行MRD检测时,都需要遵循"DFN"方法,选择较多的抗体,针对同类型白血病出现异常表达频率较高的标志进行检测。
(三)数据分析与报告模式的标准化
1.数据分析:
(1)初步设门:利用CD45/Time散点图选取液流稳定的区间;利用FSC-Height/FSC-Area和SSC-Height/SSC-Area(或FS-INT/FS-PEAK和SS-INT/SS-PEAK)散点图去除黏连体;利用FSC-Area/SSC-Area散点图去除碎片;先利用特定系列广泛表达的抗原/SSC-Area散点图圈定该系列所有细胞区域,如CD19/SSC、cyCD3/SSC、CD117/SSC圈定所有B淋巴细胞、T淋巴细胞群体及幼稚髓细胞群体,用CD38/CD138散点图圈定所有浆细胞群体;再利用CD19/CD45、cyCD3/CD45、CD117/CD45去除非特异性细胞。
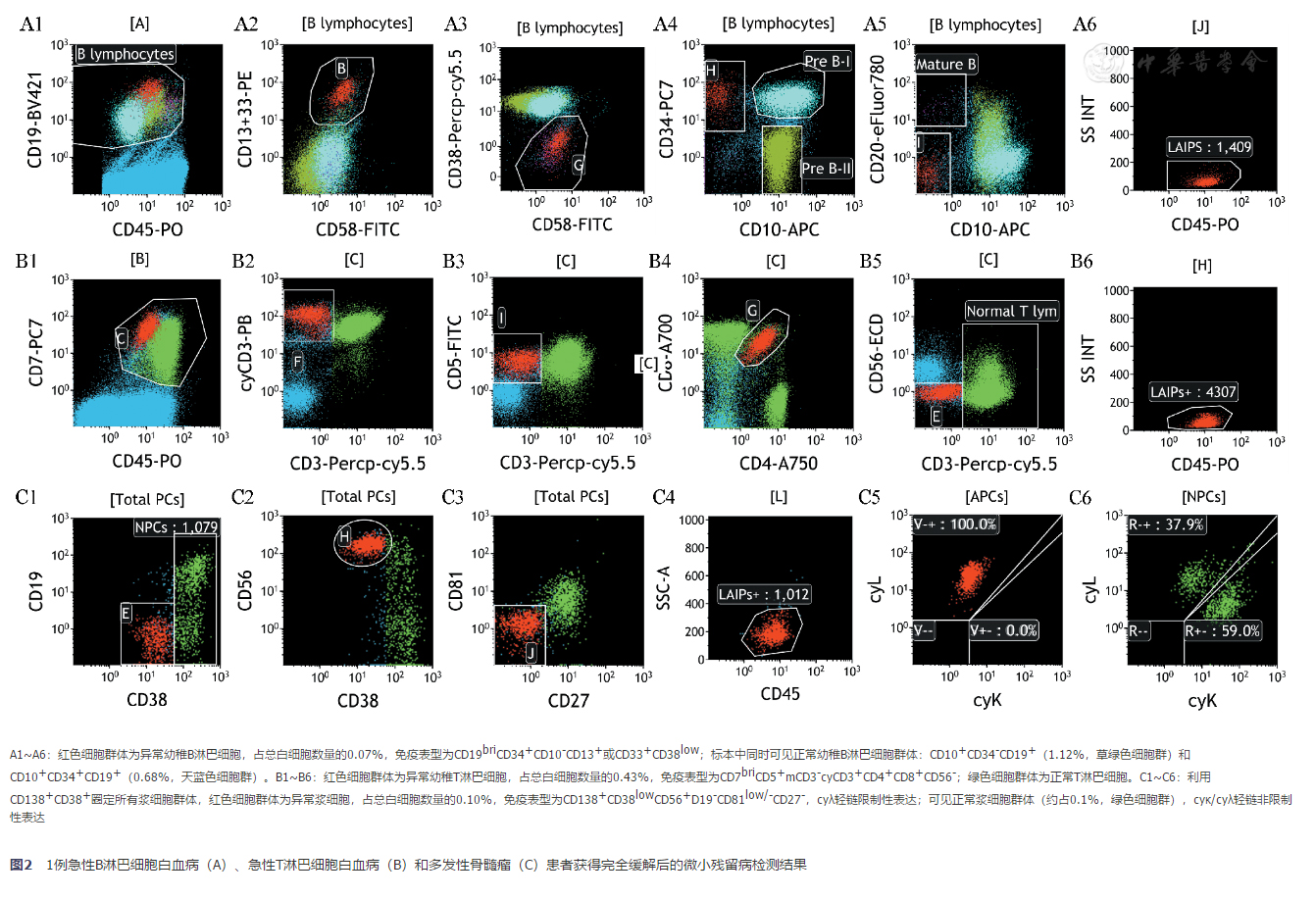
(2)MRD表型的鉴别:在分析MRD数据时,除了分析初发时的LAIP外,还应鉴别所有"DFN"的细胞群体。初发时若发现多个白血病细胞克隆,即使有些克隆含量很少,在治疗过程中也应该同时跟踪,观察克隆的变化。一般只有部分抗原在治疗过程中会发生改变,特别是系列成熟度相关抗原,其余抗原则稳定表达,因此同一患者如存在多种LAIP,尽可能同时监测,可避免出现假阴性结果[7]。利用一系列抗原表达圈定残留白血病细胞群体后,应该再次进行反向设门,将异常细胞群显示在CD45/SSC、FSC/SSC、FSC-Height/FSC-Area等散点图中,观察细胞群体是否集中、分析结果是否准确。图1和图2所示为AML、T-ALL、B-ALL和骨髓瘤各1例患者治疗后检测MRD的结果。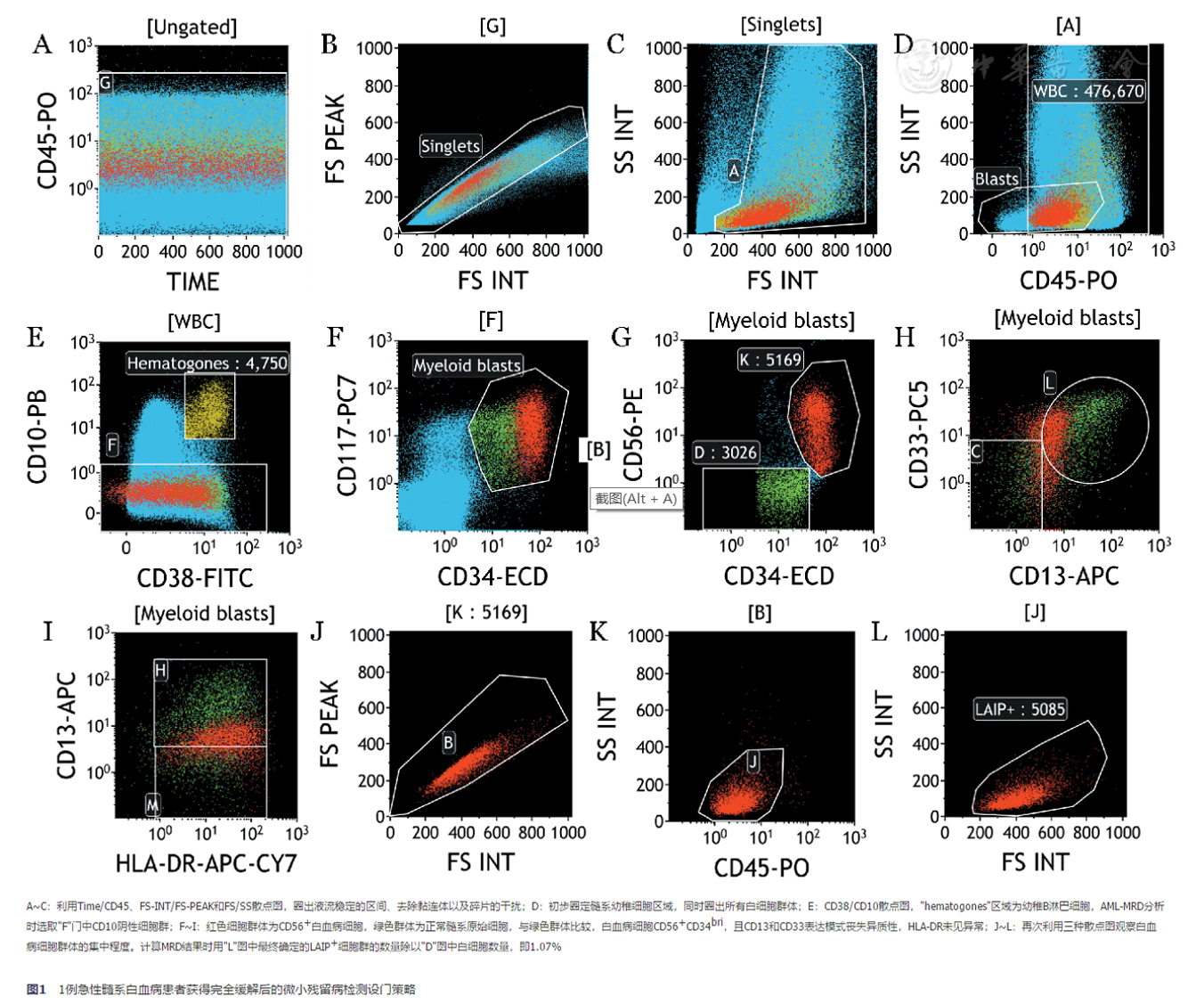
图1 1例急性髓系白血病患者获得完全缓解后的微小残留病检测设门策略
(3)MRD水平的计算:计算MRD定量结果的方法为异常细胞占所有白细胞或有核细胞的百分比。当异常细胞不表达CD45时(如PCN或部分B-ALL细胞),白细胞数量应包含CD45阳性白细胞和阴性区的异常细胞,该计算方法不受红细胞裂解效果和碎片的影响。有核细胞不仅包含白细胞数量,也包括骨髓中的其他有核成分如有核红细胞,该计算方法受红细胞裂解效果影响较大,裂解过头则分母偏小,裂解不彻底则分母偏大。利用有核细胞总数作为分母时,建议在反应体系中加入核酸染料(如Syto-16)以准确圈定有核细胞群体,防止成熟红细胞以及碎片等非有核成分造成分母偏大。
2.报告内容及报告形式:
(1)报告内容:①检测报告中首先包含患者及标本信息,标本如有凝块,特殊原因未能及时检测或稀释等情况均应注明。②MRD结果报告为"阳性"或"利用所选抗体组合,本次检测未识别出免疫表型异常细胞"。③MRD如果为阳性,应描述报告所见异常细胞数量及异常细胞占白细胞或有核细胞百分比,并描述异常细胞的免疫表型,以对应的正常造血细胞为参照,描述异常细胞的各种抗原表达与否以及表达强度,散射光(FSC和SSC)信号有异常时也应提示。④报告获取的白细胞或有核细胞总数。同时报告方法学LOD和(或)LLOQ。⑤报告中应出具一系列散点图,显示异常(亮色突出)和正常细胞群体的抗原表达及散射光信号。⑥其他内容:MRD报告中应显示抗体组合(包括荧光素)、样本制备方法以及仪器型号。注明红细胞裂解方法,以及有核细胞计数时是否利用核酸染料染色等。尽可能描述骨髓中正常细胞组分,以反映骨髓重建状态。
(2)报告形式示例:
以图1所示的病例为例,报告内容如下:
患者及标本信息:略。
仪器型号:NAVIOS(Beckman-Coulter公司,3激光10色)。
样本制备方法:全骨髓溶血法(氯化铵溶血素:实验室自行配制)。
MRD检测选用抗体组合:CD38/CD56/CD34/CD33/CD117/CD13/HLA-DR/CD10/CD45(标记荧光素种类见图中标示)。
散点图:见图1。
结果:共获取476 670个白细胞(LOD=30/476 670×100%=0.006%,LLOQ=50/476 670×100%=0.01%),异常细胞占1.07%(5 085个)。
异常免疫表型:CD117+CD34briCD38lowCD56+CD45low,CD13和CD33表达丧失异质性,为异常髓系原始细胞(见上图红色细胞群体)。
其他:可见正常CD10+幼稚B淋巴细胞(1.0%,上图黄色细胞群)和正常CD34+CD117+髓系原始细胞(0.63%,上图绿色细胞群)。
结论:MRD阳性
参加共识讨论的专家:北京大学人民医院(刘艳荣、王亚哲);天津医科大学总医院(邵宗鸿);上海交通大学医学院附属瑞金医院(翁香琴);解放军总医院(王莉莉);中国医学科学院血液病医院(王慧君);上海市临床检验中心(徐翀);南方医科大学南方医院(冯茹);第三军医大学新桥医院(张曦);兰州大学第二医院(张连生);河南省人民医院(翟亚萍);首都医科大学宣武医院(万岁桂);四川大学华西医院(朱焕玲);苏州大学附属第一医院血液病研究所(朱明清);山西肿瘤研究所免疫研究室(苏文);河北燕达医院陆道培血液肿瘤中心(王卉);江苏省人民医院(吴雨洁);深圳市人民医院(吴铭);华中科技大学同济医学院附属协和医院(郑金娥);中国中医科学院西苑医院(许勇钢);河北医科大学附属第二医院(杨静慈);山西医科大学第二医院(王宏伟);中国医科大学附属第一医院(李艳);广州军区广州总医院(李力);云南省第一人民医院(沈晓梅);福建医科大学附属协和医院(李乃农);第四军医大学附属西京医院(朱华锋);贵州医科大学附属医院(马莉);安徽省立医院(王兴兵);昆明医科大学第一附属医院(杨策尧);宁夏医科大学附属医院(杨芝红)
(执笔:翁香琴、王莉莉)
(主审:刘艳荣)